
Abstract
Key message
We constructed a high-density genetic linkage map comprising 4,593 SLAF markers using specific-locus amplified fragment sequencing and identified six quantitative trait loci for pod dehiscence resistance in soybean.
Abstract
Pod dehiscence is necessary for propagation in wild soybean (Glycine soja). It is a major component causing yield losses in cultivated soybean, however, and thus, cultivated soybean varieties have been artificially selected for resistance to pod dehiscence. Detecting quantitative trait loci (QTLs) related to pod dehiscence is required for molecular marker-assisted selection for breeding new varieties with pod dehiscence resistance. In this study, we constructed a high-density genetic linkage map using 260 recombinant inbred lines derived from the cultivars of Heihe 43 (pod-indehiscent) (ZDD24325) and Heihe 18 (pod-dehiscent) (ZDD23620). The map contained 4953 SLAF markers spanning 1478.86 cM on 20 linkage groups with an average distance between adjacent markers of 0.53 cM. In total, six novel QTLs related to pod dehiscence were mapped using inclusive composite interval mapping, explaining 7.22–24.44% of the phenotypic variance across 3 years, including three stable QTLs (qPD01, qPD05-1 and qPD08-1), that had been validated by developing CAPS/dCAPS markers. Based on the SNP/Indel and significant differential expression analyses of two parents, seven genes were selected as candidate genes for future study. The high-density map, three stable QTLs and their molecular markers will be helpful for map-based cloning of pod dehiscence resistance genes and marker-assisted selection of pod dehiscence resistance in soybean breeding.
Similar content being viewed by others

Introduction
Pod dehiscence is necessary for spreading seeds of wild plant species that bear seeds in pods and allows wild plants to introduce their progeny into a broad range of environments (Fuller 2007). However, in cultivated crops, seeds from plants that undergo pod dehiscence cannot be harvested, resulting in serious yield losses. During planting and harvesting, resistance to pods dehiscence prior to harvest is critical (Hancock 2004; Harlan et al. 1973). Therefore, during soybean domestication, pod dehiscence resistance was artificially selected as an important trait (Hideyuki et al. 2012). Although researchers intensively screen for and avoid pod dehiscence during crop domestication, pod dehiscence before harvest is still a problem in soybean breeding (Christiansen et al. 2002). Soybean breeding has made great progress in incorporating resistance to pod dehiscence, but little is known about the genetics of pod dehiscence in soybean (Funatsuki et al. 2014). The main limiting factor for genes conferring resistance to pod dehiscence is low marker coverage. This leads to large intervals for detecting and mapping QTLs related to pod dehiscence; a large number of predicted candidate genes are contained within these intervals (Zhang et al. 2016).
The rapid development of soybean genomics research has greatly enhanced the speed and the accuracy of QTL mapping for many important agronomic traits. The completion of the soybean reference genome, Williams 82, has accelerated the construction of a high-density genetic linkage map, allowing for more accurate QTL mapping and gene mining in soybean (Schmutz et al. 2010; Ju et al. 2017; Zhang et al. 2016). In soybean, researchers have constructed many genetic linkage maps using markers: restriction fragment length polymorphisms (RFLPs), simple sequence repeats (SSRs) and single-nucleotide polymorphisms (SNPs) (Gutierrez-Gonzalez et al. 2011; Hyten et al. 2010; Jeong 2002; Keim et al. 1990). To date, multiple studies have identified major QTLs for pod dehiscence in soybean using recombinant inbred lines (RILs) with RFLP (Bailey et al. 1997) and SSR markers (Funatsuki et al. 2006). These studies have shown that pod dehiscence is dominated by several major and minor QTLs. Due to the various genetic backgrounds in different soybean populations, pod dehiscence was found to be controlled by a single recessive major gene or multiple genes (Tsuchiya 1986, 1987; Kang et al. 2005; Funatsuki et al. 2008; Yamada et al. 2009). A major pod dehiscence QTL was discovered on chromosome 16 using RILs, and other minor QTLs were identified on chromosomes 2, 15 and 19 (Bailey et al. 1997). After fine mapping, the candidate gene Glyma16g25580 was confirmed to control pod dehiscence and was designated as Pod dehiscence 1 (Pdh1) (Funatsuki et al. 2014). Meanwhile, a NAC gene named SHAT1-5, Glyma.16g019400, was found to regulate pod dehiscence in soybean (Dong et al. 2014). Using SSR markers, three novel minor QTLs for pod dehiscence were confirmed on chromosomes 5, 10 and 14 from two sets of RILs (Kang et al. 2009).
Next-generation sequencing (NGS) has provided many sequencing technologies that have been used to construct high-density genetic maps on many populations. NGS techniques include restriction-site-associated DNA tagging sequencing (RAD-seq), genotyping-by-sequencing (GBS) and specific-locus amplified fragment sequencing (SLAF-seq) (Baird et al. 2008; Elshire et al. 2011; Sun et al. 2013). These techniques have become effective tools for QTL mapping and allow researchers to identify, sequence and genotype individuals in a population. Since NGS can directly identify DNA sequence differences with high accuracy, NGS techniques have been widely used for plant and animal genomics (Bhatia et al. 2013). SLAF-seq is a rapid, high-resolution technique based on NGS technology for broad-scale SNP genotyping, which has been extensively used in genomic analyses due to its characteristics of high molecular marker density, large marker number, even distribution, high accuracy and lack of repetitive markers (Sun et al. 2013). This technique can improve the efficiency of QTL mapping associated with important agronomic traits (Cao et al. 2017).
In this study, we selected SLAF markers based on the whole soybean genome through SLAF-seq. Using a set of 260 RILs derived from the cultivars Heihe 43 (ZDD24325) and Heihe 18 (ZDD23620) as parents, a high-density genetic linkage map covering the entire soybean genome was constructed to map QTLs for pod dehiscence. We obtained several QTLs associated with pod dehiscence. The high-density genetic map we generated will facilitate exploration of effective QTLs, and the novel QTLs detected in our population will be helpful for further research on pod dehiscence in soybean.
Materials and methods
Plant materials and phenotypic evaluation
We used 260 F2:7 recombinant inbred lines (RILs) that were developed by a single-seed descendent (SSD) method from an F2 population of the cross between the cultivars Heihe 43 (ZDD24325) and Heihe 18 (ZDD23620) as parents. The two parents and their derived RIL population were grown at the same location of Heihe Experiment Station, Heilongjiang Academy of Agricultural Sciences, from 2015 to 2017. The parents and RIL population were planted four rows per plot (5.0-cm plant spacing, 66.7-cm row spacing and 4.0-m row length). Heihe 43 is resistant to pod dehiscence and is the largest precocious cultivated variety in Heilongjiang province, developed from Heihe 18 and Heihe 23 (ZDD23625) by systematic breeding programs. Heihe 18 is sensitive to pod dehiscence. Although Heihe 18 and Heihe 43 have minimal differences among numerous agronomic traits, the pod dehiscence in Heihe 18 is extremely obvious (Han et al. 2015a).
We used a modified oven-dry method (Kang et al. 2005) to determine the phenotype of pod dehiscence. A total of 30 pods per line per year were carefully collected using scissors at the R8 stage and stored in sealed bags to prevent water evaporation. The pods were then incubated at 80 °C for 5 h in an oven, and the number of dehiscent pods was calculated. The PD was estimated using the following equation: PD = (dehiscent pods number/total pods number) × 100% (Peng et al. 1991).
Statistical analysis
One-way analysis of variance (ANOVA) was performed by the statistical package SAS version 9.1, including the frequency distribution, the mean of the RIL population, the coefficient of variation (CV), the broad-sense heritability (h2). The h2 for PD was estimated using the following equation: h2 = \({\sigma }_{\mathrm{G}}^{2}\)/\({\sigma }_{\mathrm{P}}^{2}\) (Nyquist and Baker 1991), where \({\sigma }_{\mathrm{G}}^{2}\) and \({\sigma }_{\mathrm{P}}^{2}\) are the genotypic and the phenotypic variance, respectively.
DNA extraction
Young, healthy and fresh leaves from both parent lines and all 260 RILs were collected in centrifuge tubes, frozen in liquid nitrogen, ground in a tissue grinder and then stored at − 80 °C. Total genomic DNA was extracted from every leaf sample following the modified CTAB protocol (Saghaimaroof et al. 1984). The quality and concentration of the extracted DNA were assessed by electrophoresis on 1% agarose gels and using a spectrophotometer (UV–Vis Spectrophotometer Q5000).
SLAF library construction and sequencing
We constructed a SLAF library and sequenced individuals from 260 RILs and their parents. The Williams 82 soybean reference genome sequence (Wm82.a2.v1, https://phytozome.jgi.doe.gov) was used to predict digestion, and we chose a combination of RsaI and HaeIII restriction enzymes to digest the genomic DNA. Amplified fragments ranging from 364 to 414 bp in size were defined as SLAF markers; 132,516 SLAF markers were predicted. A single-nucleotide (A) overhang was added to the obtained digested fragments. Dual-index sequencing adapters were then ligated to the A-tailed fragments. These fragments were generated using PCR and were then purified and mixed. The target fragments were isolated to generate a sequencing library. After purification and dilution, paired-end (each end 125 bp) sequencing was performed on an Illumina HiSeq platform (Illumina, Inc, San Diego, CA, USA). To evaluate the accuracy of the SLAF library, Oryza sativa L. Japonica was selected as a control for the same treatment and was used in library construction and sequencing. In comparison with the control data, the efficiency of enzyme digestion was evaluated to assess accuracy and effectiveness. Using reads clustering, SLAF markers were developed in parents and their offspring. Polymorphic SLAF markers were screened and used for further genetic map construction.
SLAF-seq data grouping and genotyping
The procedure of Sun et al. (2013) was adapted for SLAF marker grouping and genotyping of the 260 RILs as follows. The original sequencing read length of the SLAF-seq library was 125 bp. In order to ensure analysis quality, the original sequencing reads were filtered by removing reads containing: (1) barcode adapters; (2) > 10% base content (N); and (3) residue of restriction enzyme fragments. We selected reads ranging from 4 to 103 bp to analyze the data. Filtered reads were aligned to the reference genome using the Burrows–Wheeler Aligner (BWA) (Os et al. 2005), and reads with the same paired end were identified as the same SLAF marker. SLAF markers were analyzed for polymorphism and classification based on the number of alleles and the differences between sequences. SLAF markers were then mapped to the reference genome. The distributions of SLAF markers on chromosomes were used to map SLAF markers and polymorphic SLAF markers on the chromosomes.
Then, all polymorphic SLAF markers for genetic mapping were filtered using the following criteria: (1) average sequence depths of more than tenfold; (2) filtering out SLAF markers with more than five SNPs; and (3) integrity filtering. Markers were required to cover at least 70% of the genotypes in all progeny. Markers with missing data were filtered based on parental genotypes. The final polymorphic SLAF markers were used to construct a high-density genetic map.
Construction of a high-density genetic map
Polymorphic SLAF markers were aligned with the reference genome and mapped onto 20 chromosomes. We then calculated the MLOD (the modified logarithm of odds) scores between markers and filtered the markers with MLOD values lower than 5. Each chromosome was a linkage group (LG) used as a unit. HighMap software (Li et al. 2008) was used to analyze the linear arrangement of markers in the linkage group and to calculate the map distances. Finally, we obtained a high-density genetic map.
QTL mapping for pod dehiscence
According to the phenotypes in three environments, QTLs for pod dehiscence were detected using inclusive composite interval mapping (ICIM) in the R/qtl package (Broman et al. 2003). A total of 1000 permutation tests at a 95% confidence level were used to set the LOD threshold. Based on 1000 permutations, LOD = 2.5 was used to determine the presence of a putative QTL associated with a target trait in a certain genomic region.
Gene expression analysis by real-time qPCR (qRT-PCR)
Total RNAs of pods (R6 stage) were extracted from Heihe 43 and Heihe 18, respectively, using TransZol Up Plus RNA Kit (TransGen Biotech). For reverse transcription, the first-strand cDNA synthesis was performed using the TransScript First-Strand cDNA Synthesis SuperMix Kit (TransGen, China). For qRT-PCR, gene expressions were examined using cDNA templates on an Applied Biosystems 7300 Real-Time PCR System. Gene-specific primers for the candidate genes were designed using Primer3.0. The relative gene expression levels followed the \({2}^{-\Delta\Delta\mathrm{Ct}}\) method (Pfaffl 2001). The mRNA level of GmActin (Glyma18g52780) gene as a reference for normalization and three biological replicates were used for each gene.
Candidate gene prediction within QTL intervals
Sequences within QTLs were defined according to the Williams 82 soybean reference genome sequence (Wm82.a2.v1, https://phytozome.jgi.doe.gov). The functions of candidate genes were annotated using Blastx program (https://www.geneontology.org/) in Nr (nonredundant), Swiss-Prot and KOG/COG (clusters of orthologous groups). All genes were categorized by Gene Ontology (GO) annotation (https://www.geneontology.org/).
Development and application of molecular markers within QTLs
According to the different loci within QTLs, we developed CAPS (cleaved amplified polymorphic sequence) and dCAPS (derived cleaved amplified polymorphic sequence) markers and used these molecular markers to detect genotypes of the RILs. The online software dCAPS Finder 2.0 (https://helix.wustl.edu/dcaps/dcaps.html) was used to confirm all enzymes. Within qPD05-1, we chose a restriction site and developed a CAPS marker, in which enzyme Mnl I was used as a marker. The qPD01 QTL has no restriction enzyme sites, so we used the reverse complementary sequence and artificially introduced a mismatch base. With this method, we found a restriction enzyme site to develop a dCAPS marker with HpaII as a candidate marker enzyme. Indel markers were developed based on the Indel sites within qPD08-1. PCR primers were designed on both sides of the candidate CAPS/dCAPS markers and the Indel markers using Primer3 software (https://primer3.ut.ee/).
Genomic DNA from parents and 260 RILs were used as templates. The 20-μL PCR contained 100 ng of template DNA, 10 × PCR buffer, 2 mmol/L dNTPs, 2 mmol/L primers and 1 unit of Taq polymerase (Takara). The PCR amplification program was as follows: 95 °C for 4 min, 34 cycles of 95 °C denaturation for 30 s, 58 °C annealing for 40 s, 72 °C extension for 1 min and a final 72 °C extension for 10 min. PCR products were detected using 0.8% agarose gel electrophoresis. In accordance with the restriction enzyme digestion method (New England BioLabs, NEB), the 10-μL system consisted of 5 μL of PCR product, 0.2 μL 10 U/μL of enzyme, 1.5 μL of buffer (NEB, www.neb.com/) and 3.3 μL of ddH2O and was digested at 37 °C in a water bath for 40 min. The digested products of CAPS/dCAPS were detected using 7% polyacrylamide gel electrophoresis, and PCR products of Indels were denatured and then separated using 7% denaturing polyacrylamide gel electrophoresis (PAGE).
Results
Phenotypic analysis of pod dehiscence
We identified the PD of parents and the RILs in Heihe, Heilongjiang province, in 2015, 2016 and 2017 (Fig. 1 and Table 1). Heihe 18 had higher PD than Heihe 43 at 37% and 3% in 2015, 47.03% and 11.36% in 2016 and 75% and 3.33% in 2017. RIL population showed more resistance of pod dehiscence over 3 years, showing that the female Heihe 43 contributed to resistance of pod dehiscence and the positive skewness indicated a transgressive segregation toward lower PD. The broad-sense heritability of pod dehiscence was 81.3% in our population.

Frequency distributions of pod dehiscence in RILs from 2015 to 2017. The cultivars Heihe 43 and Heihe 18 indicated by the arrows designate the degree of pod dehiscence in parental lines
SLAF-seq and genotyping of RILs
The parent lines and RILs were sequenced and genotyped using SLAF-seq. The sequence depths in Heihe 43 and Heihe 18 were 39.74 × and 34.87 × , respectively, and the number of SLAF markers was 195,279 and 205,644, respectively. The average sequence depth was 12.35 × . The enzyme digestion efficiency was 89.82%. There were 398,386 controlled sequencing reads that were used to evaluate the accuracy of library construction; these reads were compared with the reference soybean genome. The alignment efficiency was 86.29%. The average Q30 of sequencing was 80.40%, and the average GC content was 38.12%. In total, we obtained 364,461 SLAF markers, of which 24,249 were polymorphic and the proportion of polymorphisms was 6.65%. After filtering and quality assessment, 4593 SLAF markers were used to construct a high-density genetic map. Chromosomal distribution of all SLAF markers and polymorphic SLAF markers was plotted according to the distribution of SLAFs on the chromosomes (Fig. 2). Compared with the soybean reference genome, we identified a total of 6834 SNPs, of which 4576 were transition type (Tri) and 2258 were the transversion type (Trv) (Table 2).

Distribution of SLAF markers and polymorphic SLAF markers on chromosomes. The abscissa is the length of the chromosome, and each yellow stripe represents a chromosome. The genomes were divided in units of 1 M. Different colors indicate the number of SLAFs, the color is deeper and the SLAFs are more. Darker regions in the figure are concentrated distributions of SLAF markers. The upper panel of the figure represents the distribution of the SLAF tag and the bottom panel of the figure represents the distribution of polymorphism of SLAF markers (with M as the unit)
Construction of a high-density genetic map in soybean
A total of 4593 polymorphic markers were mapped on 20 chromosomes based on the reference genome, and a high-density genetic map was constructed, with a total length of 1478.86 cM and an average distance between markers of 0.53 cM (Fig. 3). The mean chromosome length was 73.94 cM. Seven hundred and six markers were assigned on chromosome 3 with a genetic length of 101.35 cM, which was the longest of all chromosomes. Seventeen markers were assigned on chromosome 12 with a genetic length of 17.27 cM, which was the shortest of all chromosomes. The minimum number of markers was consistent with its physical length (Schmutz et al. 2010). The proportion of gaps < 5 cM between two markers was 95.72% (Table 3). Collinearity analysis (Fig. 4) of the position of markers and the genetic map on the genome showed that the order of markers on the 20 chromosomes was consistent with the genome, indicating high collinearity, indicating that the gene annotation within QTL intervals was reliable.

High-density linkage map. SLAF markers are distributed on 20 chromosomes. The black bars in each linkage group represent mapped SLAF-seq markers. The linkage group number is shown on the x axis, and genetic distance is shown on the y axis (cM is the unit)

Collinearity analyses of the genetic map and genome. The abscissa is the genetic distance of each linkage group; the ordinate is the physical length of each linkage group, which scatters the form of markers in the genome and genetic map collinearity. Different colors represent different chromosomes or linkage groups
QTL for pod dehiscence
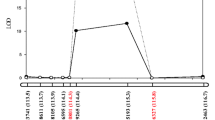
The R/qtl package was used to identify QTL associated with pod dehiscence in 3 years. In total, six novel QTLs were detected and were located on chromosomes 1, 5, 8 and 14 (Fig. 5). All of QTLs were not found in previous studies; thus, these QTLs could be considered as novel QTLs for pod dehiscence. The LOD score of these QTLs ranged from 2.61 to 11.00, and they could explain 7.22% to 24.44% of the phenotypic variation (Table 4). Of these, four QTLs (qPD01, qPD05-1, qPD05-2 and qPD08-1) explained high phenotypic variations ( > 10%). In addition, qPD01, qPD05-1 and qPD08-1, distributed on chromosomes 1, 5 and 8, respectively, could be detected in different years. Thus, these three QTLs might be the major and stable QTLs in our population. The additive effect of these QTLs was negative, which showed that the positive alleles came from the female Heihe 43 and the positive alleles improved the resistance of pod dehiscence.

Mapping of QTLs for pod dehiscence on Chr01, Chr05, Chr08 and Chr14. The curves indicate the physical position of markers against LOD score of QTL detected on chromosomes. Different lines represent different years
Candidate genes prediction and expression analysis
Based on the Williams 82 soybean reference genome, six QTL intervals contained 639 genes located on chromosomes 1, 5, 8 and 14. Currently, only 217 genes functions have been annotated by GO annotation (Fig. 6). To further select candidate genes, we compared all these sequence variations within the QTL intervals and screened out 191 SNPs and 23 Indels in 34 genes that were different between the two parents (Table 5). Of 34 genes, 32 genes were annotated their functions (Table 6). Except three genes without suitable primers to be tested, using gene-specific primers for the candidate genes (Table 7), the expression levels of the 31 candidate genes (Fig. 7) were investigated in the pods (R6 stage) collected from Heihe 43 and Heihe 18 by qRT-PCR and different levels of transcription abundance were observed among these genes. Among 31 genes, nine genes had relatively high expression levels, including Glyma.01g045700, Glyma.01g045800, Glyma.01g046000, Glyma.05g005600, Glyma.05g225900, Glyma.05g227400, Glyma.08g271900, Glyma.08g274500 and Glyma.14g195200. Seven of nine genes (Glyma.01g045800, Glyma.01g046000, Glyma.05g005600, Glyma.05g225900, Glyma.05g227400, Glyma.08g271900 and Glyma.08g274500) had significant differences between two parents.


Gene ontology (GO) annotation of genes within the QTLs. The y axis on the right shows the number of genes in each category, and the y axis on the left shows the percentage of a specific category of genes in that main category

Expression level of 31 candidate genes. Y axes indicate the ratios of relative fold expression levels. Asterisks indicate significant differences as determined by ANOVA (**P < 0.01). Relative expression was calculated based on the expression level of the GmActin gene
Development of molecular markers
According to different sites within the QTL interval, PCR amplifications were conducted using the designed primers (Table 8), and we obtained single PCR products with the same size as the target fragments. Based on alignment with Williams 82 reference sequence, there are differences between the two parents, which was consistent with the prediction.
After digestion with Mnl I, three digested products were produced theoretically by dCAPs marker in the qPD01 interval. However, one fragment of 25 bp was too small to detect; therefore, only two kinds of digestion products were detected. The DNA fragment size without digestion was 157 bp, and the allelic variation was T. Material carrying the T allele showed the pod dehiscence. After digestion, there were two fragments, 132 bp and 25 bp. The allelic variation was C, and material carrying the C allele showed the resistance of pod dehiscence. Within the qPD05-1 interval, the PCR products could be digested by HpaII and produced three digestion products theoretically. Only two kinds of digestion products could be detected due to the similar length of the two fragments after digestion. The DNA fragment size without digestion was 540 bp and the allelic variation was A that provided the resistance of pod dehiscence; the digested DNA fragment sizes were 294 bp and 246 bp and the allelic variation was G that provided the pod dehiscence. Within qPD08-1, we detected two bands: One DNA fragment size was 422 bp and the allelic variation was GTTT; the other DNA fragment size was 419 bp and the allelic variation was G that existed in the resistance of pod dehiscence. These markers were validated by using 260 RILs derived from two parents. The genotypes and phenotypes of five RILs are listed in Fig. 8 and Table 9. We found eight allele combinations in 260 RILs, and three combinations had more than ten individuals (Table 10). The identification efficiency to resistance of pod dehiscence for combination 1 with all three resistant alleles was the highest (97.4%), and both combination 2 and combination 4 with two resistant alleles were 84.2% and 82.4% respectively. The results indicated that the developed CAPS/dCAPS and Indel markers could be used to identify genotypes and predict the phenotypes of soybean varieties.

Validation of the molecular markers to QTLs (qPD01, qPD05-1 and qPD08-1). Electrophoresis results of the polymerase chain reaction (PCR) of molecular markers for Heihe 43, Heihe 18 and five recombinant inbred lines (RILs)
Discussion
QTL mapping is an effective method for analyzing quantitative traits in soybean. The quality of a genetic map has a significant impact on the resolution of QTL mapping. For a specific population, increasing marker density improves the accuracy and efficiency of a genetic map (Gutierrez-Gonzalez et al. 2011; Yu et al. 2011; Zou et al. 2012). Nevertheless, both wild and cultivated soybeans exhibit high linkage disequilibrium (LD) (cultivated soybean: ~ 150 kb; wild soybean: ~ 75 kb), and the average genetic distance is greater than in other crops (Lam et al. 2010). Therefore, a high-density genetic map is needed to improve the efficiency and accuracy of QTL mapping in soybean and can be useful for molecular marker-assisted selection (Cao et al. 2017; Staff 2014).
SLAF-seq is an efficient method for identifying QTLs and genotyping based on NGS. This technique is advantageous for large numbers of individuals due to its convenient library preparation procedure (Elshire et al. 2011). Compared with SLAF-seq, genome resequencing provides a more accurate view of the genome and captures large variations as well as small ones that may be missed by SLAF-seq. However, SLAF-seq is relatively low cost and can be used for mapping in large-scale populations (Li et al. 2014; Sun et al. 2013). SLAF-seq provides the best balance between sequencing cost and sequencing depth. SLAF-seq also obtains genome-wide variation sites at a lower cost and provides for researchers to select functional polymorphic markers, construct high-density genetic maps and further identify functional genes of important agronomic traits (Wei et al. 2017). This method has been extensively used to construct high-density genetic maps and identify major QTLs related to complex traits in several plants (Xia et al. 2015; Xu et al. 2015; Han et al. 2015a, b; Qin et al. 2015; Su et al. 2016). Pod dehiscence is one of the most important agronomic traits in crops and is directly related to yield. Therefore, in many crops, including soybean, it is vital to identify the QTLs related to pod dehiscence and the markers tightly linked to pod dehiscence. In soybean, several studies have found QTLs for pod dehiscence located on chromosome 16 and have performed fine mapping using different plant materials (Bailey et al. 1997; Dong et al. 2014; Funatsuki et al. 2014; Gao and Zhu 2013; Hideyuki et al. 2012; Kang et al. 2009), but only two genes for these pod dehiscence QTLs have been cloned. Therefore, we detected novel QTLs associated with pod dehiscence in soybean using SLAF-seq and predicted candidate genes within QTLs based on the high-density genetic maps, which could be applied for identifying new QTLs for pod dehiscence and other valuable agronomic traits. The QTLs for pod dehiscence also provided promising candidate genes for further characterization.
Constructing a high-density genetic map
Because of multiple generations of self-fertilization during the process of development of RIL populations, the observed recombination of RIL populations is greater than that of the corresponding F2 population. The increased recombination for a reference population that consists of RILs, when compared to an F2 reference population improves the accuracy and efficiency of the map. Moreover, in map-based cloning, secondary mapping populations are usually mapped after primary populations; however, doing so is laborious and time-consuming. In this study, we constructed a high-density genetic map and sequenced the whole genome in soybean RIL7 using SLAF-seq. High-quality markers were evenly distributed on 20 chromosomes. Recently, Cao et al. (2017) and Li et al. (2014) used SLAF-seq to construct high-density soybean genetic maps using 236 RILs and 200 RILs, respectively. These two genetic maps contained 3255 and 3541 SLAF markers, respectively, and the average genetic distances between two markers were 0.66 cM and 0.72 cM, respectively. Researchers have shown that increasing population number and marker density can significantly improve the efficiency of QTL mapping (Li et al. 2014). In our study, we used 260 RILs to construct a high-density genetic map containing 4593 SLAF markers. Compared with the genetic maps of Cao et al. (2017) and Li et al. (2014), the genetic distance between two adjacent SLAF markers was reduced to 0.53 cM in our map. Each chromosome contained 230 markers on average, and the shortest genetic distance between adjacent markers was 0.11 cM. Our high-density soybean genetic map may be due to the large size of the population, the increased number of SLAF markers and narrowed gaps among markers, which showed that our genetic map is an effective reference for soybean and can be used for further genetic mapping and prediction of pod dehiscence-related genes.
QTLs for pod dehiscence
The trait of pod dehiscence has considerable genetic diversity in soybean cultivars (Caviness 1965; Tsuchiya 1986; Helms 1994; Romkaew and Umezaki 2006; Yamada et al. 2009). Tsuchiya (1986) predicted that three genes control pod dehiscence. Several studies have identified two major genes controlling pod dehiscence, named qPDH1 and SHAT1-5, which are located on chromosome 16 (Bailey et al. 1997; Dong et al. 2014; Funatsuki et al. 2014; Gao and Zhu 2013; Hideyuki et al. 2012; Kang et al. 2009). Other minor QTLs related to pod dehiscence are located on chromosomes 2, 5, 10, 14, 15 and 19 (Bailey et al. 1997; Kang et al. 2009). In China, Luo et al. (2012) used 112 RILs and the composite interval mapping method and reported a QTL controlling pod dehiscence located in chromosome 6 named qPDH6-1.
Kang et al. (2009) identified seven minor QTLs for pod dehiscence using two RIL populations. Compared to this genetic map, our genetic map was constructed using a specific RIL population, which allowed us to construct a more efficient QTL map for a given phenotypic trait. We identified six novel QTLs for pod dehiscence on chromosomes 1, 5, 8 and 14 that were not detected in previous studies (Table 2). Of these six QTLs, qPD05-1 and qPD05-2 were located on chromosome 5, the same chromosome as a previously reported minor QTL (Satt 385) (Kang et al. 2009). In the USDA (United States Department of Agriculture) map, qPD05-1 is located between Sat_374 and BARCSOYSSR_05_1304, and qPD05-2 is located between BARCSOYSSR_05_0375 and BARCSOYSSR_05_0374. Interestingly, qPD05-1 and qPD05-2 were not at the same position as QTL Satt 385. Meanwhile, qPD05-1 could be found across 2 years (2015, 2016), of which the LOD and the phenotypic variation reached maximum values of 11.19% and 15.14%, respectively. Therefore, this QTL could be identified as a major, stable QTL for pod dehiscence. The other QTL, qPD14, was located on chromosome 14, the same chromosome as a previously reported minor QTL (Satt 577) (Kang et al. 2009). Similar in the previous case, qPD14 was located between Satt 063 and Sat_424. These two QTLs were not at the same position at chromosome 14.
As given in Table 2, the QTLs qPD01 and qPD08-1 could also be detected in different environments with phenotypic variation of 24.44% and 19.61%, respectively. Therefore, in our population, qPD01, qPD05-1 and qPD08-1 might be the major QTLs for pod dehiscence. qPD05-2, qPD08-2 and qPD14 were detected in a single environment; the phenotypic variation explained by qPD05-2 was 17.57%, qPD08-2 was 8.37%, and qPD14 was 7.22%. QTLs detected in the progeny could be considered stable in different environments. Therefore, these QTLs and closely linked SLAF markers can be used for molecular marker-assisted selection in soybean.
It is interesting to note that the previously reported major QTLs on chromosome 16 were not identified in our study in all environments; this result might be explained by the different genetic background between the parents of the mapping populations; the same phenomenon was found in the other trait (Li et al. 2014). The QTLs that have been identified for pod dehiscence QTL such as Pdh1 (Funatsuki et al. 2014) may be present in elite populations developed from crossing parents that are used for commercial soybean varieties including the parent we used Heihe 43, but we did not map this locus in our population. However, the QTLs that we have identified by crossing two parents that also used for developing commercial soybean varieties may have different mechanism of pod dehiscence resistance. Heihe 43 is the largest planted variety in Heilongjiang province of the People’s Republic of China at present; therefore, the breeding value of the QTLs we identified for pod dehiscence resistance can be contributed to genetic research of pod dehiscence in soybean.
Gene annotation and putative candidate genes for pod dehiscence
We predicted candidate genes within a narrow region between two adjacent markers based on high-density genetic maps and the high-quality genome sequences of Williams 82. Gene prediction depends on high accuracy and collinearity between the genetic map and the reference genome (Dan et al. 2016). The high collinearity of the markers and genetic map in our map allowed us to identify gene candidates related to pod dehiscence within QTLs (Fig. 4). Gene annotation within six QTLs was performed using Nr, Swiss-Prot and KOG/COG databases. These QTLs contained 34 protein-encoding genes, of which 32 have GO annotations (Table 6). In order to obtain more information of candidate genes in pod dehiscence, the expression profiles of 31 genes (Fig. 7) between Heihe 43 and Heihe 18 were analyzed by qRT-PCR. Among 31 genes, nine genes had relatively high expression levels, and seven of nine genes had significant differences between two parents, including Glyma.01g045800, Glyma.01g046000, Glyma.05g005600, Glyma.05g225900, Glyma.05g227400, Glyma.08g271900 and Glyma.08g274500. Previous studies have suggested that some enzymes control cell wall degradation (Christiansen et al. 2002) and synthesis (Romkaew et al. 2008). In our study, four genes involved some enzymes including fatty acid/sphingolipid desaturase (Glyma.01G046000), DNA methyltransferase (Glyma.05G005600), NADP-dependent malic enzyme (Glyma.05G227400) and oxidoreductase (Glyma.08G274500), but the function of these genes needs to be verified. Our results provided information for future study.
In this study, we constructed a high-density genetic map containing 4593 SLAF markers using 260 RILs. Our finding showed that this map was accurate and efficient for QTL mapping. We identified three stable QTLs associated with pod dehiscence resistance in three environments. These novel and stable QTLs not only contribute to study of the genetic mechanism of pod dehiscence but also facilitate molecular marker-assisted selection to increase production in soybean.
Author contribution statement
JH conducted the data analysis, QTL mapping, genomic comparative analysis and wrote the manuscript. DH and HY conducted the RIL populations and phenotyping. YG, ZW and YT provided advice on experimental implementation. LQ conceived and supervised the project, reviewed and revised the manuscript.
References
Bailey MA, Mian MAR, Carter TE, Ashley DA, Boerma HR (1997) Pod dehiscence of soybean: Identification of quantitative trait loci. J Hered 88:152–154
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376
Bhatia D, Wing RA, Singh K (2013) Genotyping by sequencing, its implications and benefits. Crop Improv 40:101–111
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Cao Y, Li S, He X, Chang F, Kong J, Gai J, Zhao T (2017) Mapping QTLs for plant height and flowering time in a Chinese summer planting soybean RIL population. Euphytica 213:39
Caviness CE (1965) Effects of relative humidity on pod dehiscence in soybeans. Crop Sci 5:511–513
Christiansen LC, Dal DFP, Borkhardt B (2002) Examination of the dehiscence zone in soybean pods and isolation of a dehiscence-related endopolygalacturonase gene. Plant Cell Environ 25:479–490
Dan Z, Li H, Wang J, Zhang H, Hu Z, Chu S, Lv H, Yu D (2016) High-density genetic mapping identifies new major loci for tolerance to low-phosphorus stress in soybean. Front Plant Sci 7:372
Dong Y, Yang X, Liu J, Wang BH, Liu BL, Wang YZ (2014) Pod shattering resistance associated with domestication is mediated by a NAC gene in soybean. Nat Commun 5:3352
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Fuller DQ (2007) Contrasting patterns in crop domestication and domestication rates: Recent archaeobotanical insights from the old world. Ann Bot 100:903–924
Funatsuki H, Ishimoto M, Tsuji H, Kawaguchi K, Hajika M, Fujino K (2006) Simple sequence repeat markers linked to a major QTL controlling pod shattering in soybean. Plant Breed 125:195–197
Funatsuki H, Hajika M, Hagihara S, Yamada T, Tanaka Y, Tsuji H, Ishimoto M, Fujino K (2008) Confirmation of the location and the effects of a major QTL controlling pod dehiscence, qPDH1, in soybean. Breed Sci 58:63–69
Funatsuki H, Suzuki M, Hirose A, Inaba H, Yamada T, Hajika M, Komatsu K, Katayama T, Sayama T, Ishimoto M (2014) Molecular basis of a shattering resistance boosting global dissemination of soybean. Proc Natl Acad Sci USA 111:17797–17802
Gao M, Zhu H (2013) Fine mapping of a major quantitative trait locus that regulates pod shattering in soybean. Mol Breed 32:485–491
Gutierrez-Gonzalez JJ, Vuong TD, Zhong R, Yu O, Lee JD, Shannon G, Ellersieck M, Nguyen HT, Sleper DA (2011) Major locus and other novel additive and epistatic loci involved in modulation of isoflavone concentration in soybean seeds. Theor Appl Genet 123:1375–1385
Han DZ, Ren YL, Guo Y, Yan HR, Zhang L, Lu WC, Qiu LJ (2015a) Occurrence characteristics and molecular genetic basis of pod shattering in soybean. Hereditas 37:535–543
Han Y, Zhao X, Cao G, Wang Y, Li Y, Liu D, Teng W, Zhang Z, Li D, Qiu L (2015b) Genetic characteristics of soybean resistance to HG type 0 and HG type 1.2.3.5.7 of the cyst nematode analyzed by genome-wide association mapping. BMC Genomics 16:1–11
Hancock JF (2004) Plant evolution and the origin of crop species. CABI Publishing, Oxford
Harlan JR, Wet JMJD, Price EG (1973) Comparative evolution of cereals. Evolution 27:311
Helms TC (1994) Greenhouse and field evaluation of pod dehiscence in soybean. Can J Plant Sci 74:699–701
Hideyuki F, Makita H, Tetsuya Y, Masaya S, Seiji H, Yoshinori T, Shohei F, Masao I, Kaien F (2012) Mapping and use of QTLs controlling pod dehiscence in soybean. Breed Sci 61:554–558
Hyten DL, Choi IY, Song QJ, Specht JE, Carter TE Jr, Shoemaker RC, Hwang EY, Matukumalli LK, Cregan PB (2010) A high density integrated genetic linkage map of soybean and the development of a 1536 universal soy linkage panel for quantitative trait locus mapping. Crop Sci 50:960–968
Jeong SC (2002) Mapping tightly linked genes controlling potyvirus infection at the Rsv1 and Rpv1 region in soybean. Genome 45:592–599
Ju SL, Kim KR, Ha BK, Kang S (2017) Identification of SNPs tightly linked to the QTL for pod shattering in soybean. Mol Breed 37:54
Kang ST, Hyeunkyeung K, Inyoul B, Mounggun C, Han WY, Doochull S, Sukha L (2005) Genetic analysis of pod dehiscence in soybean. Korean J Cropence 50:281–285
Kang ST, Myounghai K, Hyeunkyeung K, Myounggun C, Han WY, Inyoul B, Moonyoung K, Kyujung V, Sukha L (2009) Population-specific QTLs and their different epistatic interactions for pod dehiscence in soybean [Glycine max (L.) Merr.]. Euphytica 166:15–24
Keim P, Diers BW, Olson TC, Shoemaker RC (1990) RFLP mapping in soybean: association between marker loci and variation in quantitative traits. Genetics 126:735
Lam HM, Xu X, Liu X, Chen W, Yang G, Wong FL, Li MW, He W, Qin N, Wang B (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42:1053–1059
Li R, Li Y, Kristiansen K, Wang J (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24:713–714
Li B, Ling T, Zhang J, Long H, Han F, Yan S, Wang L, Zheng H, Sun J (2014) Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC Genomics 15:1086
Luo RY, Gong PT, Zhao DG, Liu XY (2012) QTL mapping and analysis of traits related to pod dehiscence in soybean. Legume Genomics Genet 3:14–20
Nyquist WE, Baker RJ (1991) Estimation of heritability and prediction of selection response in plant-populations. Crit Rev Plant Sci 10:235–322
Os HV, Stam P, Visser RGF, Eck HJV (2005) SMOOTH: a statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor Appl Genet 112:187
Peng YH, Yuan JZ, Yang GB (1991) Primary analysis of soybean shattering in Chinese yellow river and Huai river soybean adaptation region. Soybean Sci 10:285–290
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:2002–2007
Qin D, Dong J, Xu F, Guo G, Ge S, Xu Q, Xu Y, Li M (2015) Characterization and fine mapping of a novel barley stage green-revertible albino gene (HvSGRA) by bulked segregant analysis based on SSR assay and specific length amplified fragment sequencing. BMC Genomics 16:838
Romkaew J, Umezaki T (2006) Pod dehiscence in soybean: assessing methods and varietal difference. Plant Prod Sci 9:373–382
Romkaew J, Nagaya Y, Goto M, Suzuki K, Umezaki T (2008) Pod dehiscence in relation to chemical components of pod shell in soybean. Plant Prod Sci 11:278–282
Saghaimaroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Staff TPO (2014) Correction: A high-density genetic map for soybean based on specific length amplified fragment sequencing. PLoS ONE 9:e104871
Su J, Pang C, Wei H, Li L, Bing L, Wang C, Song M, Wang H, Zhao S, Jia X (2016) Identification of favorable SNP alleles and candidate genes for traits related to early maturity via GWAS in upland cotton. BMC Genomics 17:687
Sun X, Liu D, Zhang X, Li W, Liu H, Hong W, Jiang C, Guan N, Ma C, Zeng H (2013) SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 8:e58700
Tsuchiya T (1986) Studies on shattering resistance in soybean breeding. Rep Hokkaido Prefect Agric Exp Station 58:1–53
Tsuchiya T (1987) Physiological and genetic analysis of pod shattering in soybeans. Jarq 21:166–175
Wei Q, Fan GY, Liu DD, Zhang HL, Wang XW, Jian W, Xu ZS (2017) Construction of a high-density genetic map and the X/Y sex-determining gene mapping in spinach based on large-scale markers developed by specific-locus amplified fragment sequencing (SLAF-seq). BMC Genomics 18:276
Xia C, Chen LL, Rong TZ, Li R, Xiang Y, Wang P, Liu CH, Dong XQ, Liu B, Zhao D (2015) Identification of a new maize inflorescence meristem mutant and association analysis using SLAF-seq method. Euphytica 202:35–44
Xu F, Sun X, Chen Y, Huang Y, Tong C, Bao J (2015) Rapid identification of major QTLs associated with rice grain weight and their utilization. PLoS ONE 10:e0122206
Yamada T, Funatsuki H, Hagihara S, Fujita S, Tanaka Y, Tsuji H, Ishimoto M, Fujino K, Hajika M (2009) A major QTL, qPDH1, is commonly involved in shattering resistance of soybean cultivars. Breed Sci 59:435–440
Yu H, Xie W, Wang J, Xing Y, Xu C, Li X, Xiao J, Zhang Q (2011) Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS ONE 6:e17595
Zhang D, Li H, Wang J, Zhang H, Hu Z, Chu S, Lv H, Yu D (2016) High-density genetic mapping identifies new major loci for tolerance to low-phosphorus stress in soybean. Front Plant Sci 7:372
Zou G, Zhai G, Feng Q, Yan S, Wang A, Zhao Q, Shao J, Zhang Z, Zou J, Han B (2012) Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J Exp Bot 63:5451
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFD0100304) and the National Science and Technological innovation Program of China (2060302–2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Communicated by Istvan Rajcan.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Han, J., Han, D., Guo, Y. et al. QTL mapping pod dehiscence resistance in soybean (Glycine max L. Merr.) using specific-locus amplified fragment sequencing. Theor Appl Genet 132, 2253–2272 (2019). https://doi.org/10.1007/s00122-019-03352-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03352-x