
Abstract
This paper is concerned with a novel statistical–genetic approach for the construction of linkage maps in populations obtained from reciprocal translocation heterozygotes of barley (Hordeum vulgare L.). Using standard linkage analysis, translocations usually lead to ‘pseudo-linkage’: the mixing up of markers from the chromosomes involved in the translocation into a single linkage group. Close to the translocation breakpoints recombination is severely suppressed and, as a consequence, ordering markers in those regions is not feasible. The novel strategy presented in this paper is based on (1) disentangling the “pseudo-linkage” using principal coordinate analysis, (2) separating individuals into translocated types and normal types and (3) separating markers into those close to and those more distant from the translocation breakpoints. The methods make use of a consensus map of the species involved. The final product consists of integrated linkage maps of the distal parts of the chromosomes involved in the translocation.
Similar content being viewed by others


Introduction
At present barley (Hordeum vulgare L.) is the fourth most important cereal crop, after maize, wheat and rice. Barley is a diploid (2n = 14) self-pollinating species which can be used as a model plant for other Triticeae species providing an excellent system for genome mapping and map-based analyses (Costa et al. 2001). Well-established linkage maps containing morphological, chromosomal, protein and molecular markers are indispensable tools for genetic analysis and for analysing DNA sequence variation between the crops and its relative accessions and species. In barley, common molecular marker technologies include restriction digest based RFLPs (Restriction Fragment Length Polymorphisms; Graner et al. 1991; Heun et al. 1991; Kleinhofs et al. 1993), and PCR-based markers like AFLPs (Amplified Fragment Length Polymorphism; Waugh et al. 1997), STS (Sequence Tagged Site; Olson et al. 1989), SSRs (Simple Sequence Repeat; Liu et al. 1996; Ramsay et al. 2000; Pillen et al. 2000; Li et al. 2003), and recently, DArT (Diversity Arrays Technology), a technique based on DNA hybridisation (Jaccoud et al. 2001; Wenzl et al. 2004). The more recent SNPs (Single Nucleotide Polymorphisms) have become popular thanks to a high density within the genome, easy development from sequence data and high reproducibility. Wenzl et al. (2006) constructed a high-density linkage map which will be used as a reference for future mapping work. The map was based on datasets from multiple populations, comprising 2,935 loci (2,085 DArT, 850 SSR, RFLP and/or STS markers) and spans 1,161 cM. Recently, Varshney et al. (2007) produced a high-density linkage map, using SSR marker data from six mapping populations. A total of 775 microsatellite loci from 688 primer pairs were used to cover 1,068 cM of the barley genome.
Major complications in the construction of genetic maps arose in populations containing reciprocal translocation heterozygotes (Lehmensiek et al. 2005). Reciprocal translocations involve the exchange of two terminal segments between two non-homologous chromosomes. Due to reciprocal translocations recombination between loci around the translocation breakpoints will be suppressed. Consequently, markers in these regions become ‘pseudo-linked’, i.e. linkage is detected between markers lying on different chromosomes. Reciprocal translocations are induced spontaneously or artificially by ionizing radiation, mutagens or by transposons and can be confirmed by aberrant chromosome and banding morphology, by establishing quadrivalent formation in metaphase I complements, and semi-sterility of pollen in translocation heterozygotes, and by linkage analysis (Jáuregui et al. 2001). Translocations are well documented in various crops, including rye (Alonso-Blanco et al. 1993; Benito et al. 1994; Catarino et al. 2006), soybean (Mahama and Palmer 2003), Prunus (Jáuregui et al. 2001), Lens (Tadmor et al. 1987) and pea (Kosterin et al. 1999). Many bread wheat cultivars contain the 1RS-1BL wheat–rye chromosome translocation, introduced to provide resistance to pests and diseases (Mago et al. 2002; Sharma et al. 2009).
In barley, spontaneous reciprocal translocations were described by Konishi and Linde-Laursen (1988) who investigated 1,240 barley landraces and varieties and 120 wild barley lines, using test-cross and Giemsa chromosome banding analyses. Of the 1,240 landraces and varieties, four carried translocations involving chromosomes 2H and 4H with breakpoints near or at the centromere. Of the 120 wild barley lines, three carried translocations between chromosomes 2H and 4H, 3H and 5H, 3H and 6H, respectively. ‘Albacete’ is a barley variety that it is known to have a reciprocal chromosomal translocation. Recent results suggest that chromosomes 1H and 3H are involved in this translocation (Lacasa-Benito et al. 2005).
The consensus map of barley (Wenzl et al. 2006) was used as a reference. Preliminary linkage analysis of a doubled haploid population obtained from a cross between ‘Albacete’ and the non-translocation barley variety Barberousse shows that linkage groups 1H and 3H are indeed involved in the translocation. In order to detect unexpected relationships between markers at those chromosomes we apply a principal coordinate analysis which visualises marker positions in a three-dimensional graph. The next step of our analysis is based on the cytogenetical theory that (1) during meioses two types of viable gametes are produced: a “normal” type (with a balanced set of non-translocation chromosomes) and a “translocated” type (with a balanced set of translocated chromosomes), and (2) that around the translocation breakpoints recombination is suppressed (Burnham 1962; Sybenga 1975). The crucial step consists of dividing the doubled haploid population into two subpopulations according to the origin of the alleles at the translocation breakpoints: Barberousse alleles refer to the “normal” type and Albacete alleles refer to the “translocated” type. The position of the translocation breakpoint is found by a graphical display of recombination frequencies versus positions on the consensus map. At this stage we discuss the problem of extreme segregation distortion around the translocation breakpoints in the two subpopulations, which is solved by removing markers around the translocation breakpoints with severe segregation distortion. Linkage maps for the various chromosome arms are then obtained for each of the subpopulations separately. The final step is integrating the linkage maps of the corresponding arms of linkage groups 1H and 3H of the two subpopulations.
Materials and methods
Plant material
Two 6-row parental barley varieties, ‘Albacete’ and ‘Barberousse’, were crossed and anther culture lines were produced from the F1 from which 231 spontaneous doubled haploids were obtained at Estación Experimental de Aula Dei, Zaragoza, Spain. Seed from each doubled haploid line was retained as a reference stock and used to grow the plants for DNA extraction. More than 50 years ago, ‘Albacete’ was found by Enrique Sánchez-Monge at the Estación Experimental Aula Dei, Spanish Research Council, as an inbred line in a local heterogeneous landrace population from Albacete (province of Spain). It is a six-row barley variety, drought tolerant with a stable grain yield. For these properties, it has been grown for decades on up to 1 million ha/year, especially in drought-prone areas. ‘Barberousse’ is a six-row winter barley variety released in France in 1977. It was obtained from the cross [259 711/(Hatif de Grignon/Ares)]/Ager. Although it is sensitive to drought, this variety is known for its good productivity and easy adaptation.
DNA isolation
Genomic DNA of the DHs was extracted using two different protocols. For genomic and EST-derived SSR markers, genomic DNA was extracted from 0.1 g of fresh leaf tissue sprayed with liquid nitrogen according to the method of Doyle and Doyle (1990) with some modifications. DNA was diluted in TE buffer pH8 and preserved for a long period at a temperature of −20°C. For DArT markers the genomic DNA was extracted from fresh leaf tissue using the Qiagen DNeasy 96 Plant Kit.
Genotyping
Genotypic data were first produced for genomic and EST-derived SSR markers. We refer to the Genomic SSRs markers using the prefix (“Bmac” or “Bmag”) followed by four digits and to the EST-derived SSR markers using the prefix (“scssr”) followed by five digits. Genomic and EST-derived SSR analyses were carried out using the fluorescent fragment detection system on an ABI PRISM® 3100-Avant Genetic Analyzer using Genescan software from Applied Biosystems. For this method, the reverse primer was labelled with a fluorochrome at the 5′ end. The PCR programmes used for all amplifications are published in the UK CropNet databases (http://ukcrop.net/db.html).
Subsequently, a genome-wide scan was carried out using the Diversity Arrays Technology (DArT, http://www.diversityarrays.com). In a single assay, DArT uses microarrays to detect DNA polymorphisms at several hundred genomic loci spread over the entire genome. It is based on hybridising labelled genomic representations of individual DNA samples on a micro-array, which contains a large number of DNA fragments derived from the total genomic DNA of the species under consideration (Wenzl et al. 2004). Polymorphisms were scored as presence (=1) or absence (=0) of hybridisation to individual array elements. The locus designations of Triticarte Pty. Ltd. were used. DArT markers consisted of prefix (“bPb” or “CloneID”) followed by a number corresponding to the particular clone in the genomic representation.
Linkage analysis
For the construction of linkage maps we used the maximum likelihood (ML) algorithm in Jansen et al. (2001) which enables map construction in batch mode and the implementation in JoinMap 4.0 (van Ooijen 2006) which enables the use of many graphical tools. Linkage maps were drawn using MapChart 2.2 (Voorrips 2002).
Statistical techniques
Principal coordinate analysis (Chatfield and Collins 1980), a special form of multidimensional scaling, was used to obtain a 3D representation of the markers on the linkage groups involved in the reciprocal translocation. The similarity between markers was measured by the simple matching coefficient, which for a doubled haploid population is equal to one minus the recombination frequency. Calculations were carried with Genstat version 12 (Payne et al. 2009), using the directives FSIMILARITY and PCO.
Results
Polymorphisms between parents
We applied the following liberal rules for excluding markers: (1) markers with an unknown location on the consensus map (these were later included again when constructing our final maps); (2) markers with an ambiguous chromosome assignment on the consensus map; (3) markers with more than 5% missing data and (4) markers with low quality readings (Q value less than 77). For linkage analysis, 30 polymorphic genomic and EST-derived SSR markers and 309 polymorphic DArT were used. This amounted to an average of 48 markers per chromosome, with an average interlocus distance of 4.4 cM based on the consensus linkage map. A few regions had gaps of over 10 cM. Chromosome 4H showed strikingly low levels of polymorphisms, which is in agreement with the findings of Qi et al. (1996), Karakousis et al. (2003), Kleinhofs et al. (1993) and Wenzl et al. (2006).
Preliminary linkage map construction
We obtained six main linkage groups involving 305 markers. Using a recombination frequency threshold of 0.2, 34 markers could not be assigned to one of these linkage groups. Five linkage groups could be related to linkage groups of the consensus map (Wenzl et al. 2006): 2H (48 markers), 4H (20 markers), 5H (48 markers), 6H (49 markers) and 7H (58 markers). The remaining 82 markers had known positions on 1H and 3H of the consensus map, of which 78 formed one linkage group. Four markers, on the consensus map located at the very end of the long arm of linkage group 1H, were found to be unlinked with the 78 markers of 1H and 3H. One marker showing a consistently high nearest-neighbour stress (Van Ooijen 2006) was also excluded. Figure 1 displays one linkage map with the positions of the remaining 77 markers obtained by the maximum likelihood algorithm. Many different linkage maps were obtained by rerunning the maximum likelihood algorithm. This was a clear indication that it was not possible to obtain a linear order of the markers in an unambiguous way.

Graphical representation of a linkage map obtained for markers of linkage groups 1H and 3H according to the consensus map of Wenzl et al. (2006); markers of linkage group 1H and 3H are displayed in blue/dark grey and ochre/light grey, respectively
A spatial representation of the positions of the markers was obtained using principal coordinate analysis. Figure 2 shows a three-dimensional plot in which the points represent the positions of the markers with regard to the first three principal axes; the percentage of variance explained by the first three axes was 27.2, 15.5 and 13.9, respectively. The points have been joined by straight lines according to their position on the consensus map of Wenzl et al. (2004). Markers of linkage group 1H and 3H have been displayed in blue and ochre, respectively. Figure 2 shows that linkage groups 1H and 3H merge at one point (indicated by the circle) and that the short and long chromosome arms divert from this point, and from each other, in three-dimensional space. As a consequence, the translocation breakpoints lie within the circle.

Graphical representation of a principal coordinate analysis using the simple matching coefficient for the markers of linkage groups 1H and 3H according to the consensus map of Wenzl et al. (2006). The percentage of variance explained by the first three principal axes is 27.2, 15.5 and 13.9, respectively. The markers have been joined according to their position on the consensus map of Wenzl et al. (2006). The black circle encloses markers of linkage groups 1H and 3H that are close to the translocation breakpoints; they are separated by a small number of recombinations
Recombination around the translocation breakpoints
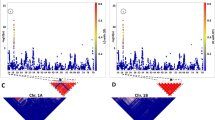
To study the recombination behaviour of markers around the translocation breakpoints we focussed on estimates of the recombination frequencies between the markers of 1H and 3H, in which the allocation of markers to linkage groups as well as their positions were based on the consensus map. In Fig. 3, these recombination frequencies were plotted against the corresponding positions on the consensus map. In Fig. 3a, we observed that markers close to the translocation breakpoints have recombination frequencies close to zero. On linkage group 1H the translocation breakpoint was situated at about 60 cM and on linkage group 3H at about 65 cM based on the consensus map. The marker pair with the smallest recombination frequency consisted of HvM20 (1H) and Bmag0136 (3H) with a recombination frequency of 0.017 (4 recombinants out of 231 DHs). Cross-sections of Fig. 3a through the positions of markers Bmag0136 and HvM20 are shown in Fig. 3b, c, respectively.

a Graphical representation of the recombination frequencies between markers of linkage group 1H and linkage group 3H versus their position on the consensus map of Wenzl et al. (2006). b Graphical representation of the recombination frequencies between markers of linkage group 1H with marker Bmag0136 of linkage group 3H versus their position on linkage group 1H of the consensus map of Wenzl et al. (2006). c Graphical representation of the recombination frequencies between markers of linkage group 3H with marker HvM20 of linkage group 1H versus their position on linkage group 3H on the consensus map of Wenzl et al. (2006)
Cytogenetical theory (see Appendix) suggests that the DH1 population should be split into two subpopulations: (1) a subpopulation containing individuals in which the alleles close to the translocation breakpoint are identical to Albacete, and (2) a subpopulation containing individuals in which the alleles close to the translocation breakpoint are identical to Barberousse. In the first subpopulation, the chromosomal arrangement will be that of Albacete (1HS-3HL, 3HS-1HL), in the second subpopulation the chromosomal arrangement will be that of Barberousse (1HS-1HL, 3HS-3HL). The separation of the 231 DH individuals into two subpopulations was based on the data of markers HvM20 and Bmag0136 (Fig. 4). First, the markers were divided into linkage groups 1H and 3H and subsequently sorted vertically according to their position within each of the linkage groups based on the consensus map. Secondly, individuals were sorted according to the origin of their allele on marker HvM20; more or less the same effect was obtained if the markers were sorted according to Bmag0136. The split of the individuals into two groups was not perfect; 53 individuals had to be excluded because they had deviating chromosomal combinations within 5 cM from the presumed translocation breakpoints. Some of these deviations concerned so-called ‘singletons’; they are likely due to observation errors or caused by gene conversions.

Graphical genotypes of individuals markers on linkage 1H and 3H according to the consensus map of Wenzl et al. (2006). Individuals have been divided into two types with regard to their alleles at the translocation breakpoints: the Albacete type (shown in red/dark grey), carrying the translocation, and the Barberousse type (shown in green/light grey), with ‘normal’ chromosome arrangement
In a doubled haploid population, markers are expected to segregate according to a 1:1 ratio. However, due to the division into subpopulations, markers near the translocation breakpoints show severe segregation distortion in each of the subpopulations. The distortion decreases as markers are further away from the translocation breakpoints. Severe segregation distortion significantly affects estimation of genetic distances, but will not seriously affect the marker order (Hackett and Broadfoot 2003). Therefore, in order to construct robust linkage maps, markers with severe segregation distortion (in this case markers with segregation ratios below 30% or above 70%) were eliminated. In both subpopulations 37 markers out of 75 were removed, leaving 14 markers for 1H and 24 markers for 3H; these markers were common in both subpopulations.
The two subpopulations were considered separately to construct independent linkage maps for the chromosomes involved in the reciprocal translocation. Subpopulation 1 (with the ‘Albacete’ allele near or at the translocation breakpoint) contained 92 DHs; Subpopulation 2 (‘Barberousse’ allele near the translocation breakpoint) contained 86 DHs. The two subpopulations were then subjected to independent linkage analyses. Finally the linkage maps of the two subpopulations were integrated. Results are shown in Fig. 5. The order of the markers for the ‘Albacete’ and the ‘Barberousse’ population are identical, but differences in distance are present.

Linkage maps of the short and long arms of linkage groups 1H and 3H obtained for the individuals of type Albacete and for type Barberousse at the translocation breakpoints. The maps of the two types have been connected by their corresponding integrated maps. The maps were obtained for markers with a segregation ratio between 0.3 and 0.7
Discussion
The methods developed in this paper provide essential tools for disentangling ‘pseudo-linkage’ between markers in a population derived from a translocation heterozygote. The methods can be implemented using standard software. In this paper a high-quality consensus map of ‘normal’ barley (used as a reference) is essential in several steps of the analysis: (1) interpretation of the three-dimensional principal coordinate plot (Fig. 2), (2) determination of the translocation breakpoints on chromosomes 1H and 3H of ‘normal’ barley (Fig. 3), and (3) assignment of markers to the respective chromosome arms (1HS, 1HL, 3HS and 3HL) in ‘normal’ barley (Fig. 4). Without a high-quality, consensus map of ‘normal’ barley, step (2) will be impossible, and steps (1) and (3) will be more difficult. The chromosome arms designated 1HS, 1HL, 3HS and 3HL become arbitrary chromosome arms.
In a preliminary linkage analysis, six main linkage groups were detected; one of them joined markers from chromosomes 1H and 3H. As indicated by an earlier study this “mixing” of markers of chromosomes 1H and 3H was attributed to the presence of a reciprocal translocation between these chromosomes. In this study the position of the translocation breakpoint could be determined at about 60 cM on chromosome 1H and 65 cM on chromosome 3H (positions refer to the consensus map of ‘normal’ barley), which correspond with the centromeric regions of these chromosomes. These positions were confirmed in this study when all available markers from 1H and 3H were used to determine in more detail which markers were involved in the translocation breakpoints. In another DH1 population obtained from a cross of ‘Albacete’ and ‘Plaisant’ (results not shown), the translocation breakpoints were found at similar positions.
In order to characterise the translocation breakpoint we identified 92 DHs as carrying the translocation and 86 DHs with the standard chromosome arrangement. Fifty-three genotypes could not be classified in either of the two subpopulations due to deviating chromosomal combinations which may be caused by scoring errors. In marker studies like these scoring errors should be avoided by using very accurate marker systems. Nevertheless, it is interesting to note that, regardless of the true nature of the 53 non-classified types, the presence of this chromosome rearrangement did not affect the success rate in the double haploid regeneration process. As a result, two different genetic linkage maps were obtained, one with the two interchanged chromosomes together (1HS·3HL + 1HL·3HS, ‘Albacete’ type) and the other with the two normal chromosomes together (1HS·1HL + 3HS·3HL, ‘Barberousse’ type).
It is expected that barley varieties carrying chromosomal segments containing the reciprocal translocation between chromosomes 1H and 3H, have an advantage under certain environmental conditions. So, for future breeding purposes, it may be worthwhile to be able to follow individuals carrying interchanged chromosomes. In our case, this is the ‘Albacete’ haplotype. This requires the development of markers close enough to the translocation breakpoints with allelic combinations that allow differentiation between translocated and non-translocated origin of the chromosomal segments around the translocation breakpoints; in our case marker haplotypes that distinguish between Albacete and all other material used in the breeding programme. Away from the translocation breakpoints, a mixing of haplotypes will occur due to recombination. In the end, this would allow the construction of a specific linkage map of the karyotype with a chromosomal rearrangement close to the translocation breakpoints.
A genetic consequence of reciprocal translocation is that linkage relationships in a translocation heterozygote are altered as recombination between loci may be significantly reduced, particularly within the interstitial segment and between genes close to the translocation breakpoint. If the translocation breakpoint is located at or near the centromeric region, the interstitial zone becomes really narrow or even null and, therefore, the recombination that may take place will be due to interference effects near the translocation breakpoint. In our data we have not found a common set of completely linked markers from chromosome 1H and 3H, although a few showed very low levels of recombination. These rare recombinants can be due to scoring errors or, what is more likely, due to the fact that the interstitial zone is practically non-existent.
The genomic SSR markers linked closely to the translocation breakpoint on chromosome 3H were identified flanking the centromere at 55 cM in the Lina × Hordeum spontaneum ‘Canada Park’ genetic map, between Bmac0067, Bmag0136 and Bmag006 in the short arm and Bmac0209 in long arm covering a genetic distance of 5 cM (Künzel and Waugh 2002). However, this region (with suppressed recombination) covered 36% of the entire chromosome length on the physical map. For chromosome 1H, no articles were found, which relate physical distance to genetic distance using genomic SSR markers along the entire chromosome; only one RFLP-based map observed this phenomenon (Künzel et al. 2000). The gene density around the translocation breakpoints can be inferred using the synteny observed for barley-rice. Mayer et al. (2009) mentioned that approximately 20% of all genes on barley chromosome 1H are located in centromeric and subcentromeric regions with very low recombination frequencies. A similar gene density can be expected for barley chromosome 3H; Smilde et al. (2001) reported a 30-fold reduction of recombination around the barley 3H centromere.
Livingstone et al. (2000) state that karyotypic rearrangements, such as reciprocal translocations, lead to high variances of inter-marker distances for markers on chromosomes involved in the reciprocal translocation away from the translocation breakpoints, in repeated attempts to construct a linkage map (see also Durrant et al. 2006). Markers close to the translocation breakpoints show small variances of inter-marker distances. However, high variances may also be caused by deficiencies in the data. The methods developed in this paper provide a novel combination of methods available in standard (statistical) software that can be used to identify the true background of ‘pseudo-linkage’ in a straightforward manner.
In conclusion, the results of this study confirm that there is a reciprocal translocation as the result of an interchange of chromosome arms between 1H and 3H. The markers found to be linked to the translocation breakpoints are HvM20 and EBmac0501 for 1H and Bmag0136, bPb-9746 and a reduced set of genomic SSR markers for 3H, which are located around the centromere. Linkage analysis revealed that alternative arrangements involving the short and long chromosome arms of 1H and 3H may be possible. However, the most frequent arrangement is 1HS·3HL + 3HS·1HL. In addition, regardless of the presence of a reciprocal translocation we have produced a linkage map for the ‘Albacete’ × ‘Barberousse’ population that will be used to identify quantitative trait loci (QTLs) for agronomic traits in future work.
References
Alonso-Blanco C, Goicoechea PG, Roca A, Giraldez R (1993) A cytogenetic map on the entire length of rye chromosome 1R, including one translocation breakpoint, three isozyme loci and four C-bands. Theor Appl Genet 85:735–744
Benito C, Llorente F, Henriques-Gil N, Gallego FJ, Zaragoza C, Delibes A, Figueiras AM (1994) A map of rye chromosome 4R with cytological and isozyme markers. Theor Appl Genet 87:941–946
Burnham CR (1962) Discussions in cytogenetics. Burgess, Minneapolis
Catarino S, Alvarez E, Campa A, Vieira R, Roca A, Giraldez R (2006) Identification and physical mapping of induced translocation breakpoints involving chromosome 1R in rye. Chromosom Res 14:755–765
Chatfield C, Collins AJ (1980) Introduction to multivariate analysis. Chapman and Hall, London
Costa JM, Corey A, Hayes PM, Jobet C, Kleinhofs A, Kopisch-Obusch A, Kramer SF, Kudrna D, Li M, Riera-Lizarazu O, Sato K, Szucs P, Toojinda T, Vales MI, Wolfe RI (2001) Molecular mapping of the Oregon Wolfe barleys: a phenotypically polymorphic doubled-haploid population. Theor Appl Genet 103:415–424
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissues. Focus 12:13–15
Durrant JD, Gardunia BW, Livingstone KD, Stevens MR, Jellen EN (2006) An algorithm for analyzing linkages affected by heterozygous translocations: QuadMap. J Hered 97:62–66
Graner A, Jahoor A, Schondelmaier J, Siedler H, Pillen K, Fischbeck G, Wenzel G, Herrmann RG (1991) Construction of an RFLP map of barley. Theor Appl Genet 83:250–256
Hackett CA, Broadfoot LB (2003) Effects of genotyping errors, missing values and segregation distortion in molecular marker data on the construction of linkage maps. Heredity 90:33–38
Heun M, Kennedy AE, Anderson JA, Lapitan NLV, Sorrells ME, Tanksley SD (1991) Construction of a restriction fragment length polymorphism map for barley (Hordeum vulgare). Genome 34:437–447
Jaccoud D, Peng K, Feinstein D, Kilian A (2001) Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res 29:e25
Jansen J, de Jong AG, van Ooijen JW (2001) Constructing dense genetic linkage maps. Theor Appl Genet 102:1113–1122
Jáuregui B, Vicente MC, Messeguer R, Felipe A, Bonnet A, Salesses G, Arús P (2001) A reciprocal translocation between ‘Garfi’ almond and ‘Nemared’ peach. Theor Appl Genet 102:1169–1176
Karakousis A, Gustafson JP, Chalmers KJ, Barr AR, Langridge P (2003) A consensus map of barley integrating SSR, RFLP, and AFLP markers. Aust J Agric Res 54:1173–1185
Kleinhofs A, Kilian A, Saghai Maroof MA, Biyashev RM, Hayes P, Chen FQ, Lapitan N, Fenwick A, Blake TK, Kanazin V, Ananiev E, Dahleen L, Kudrna D, Bollinger J, Knapp SJ, Liu B, Sorrells M, Heun M, Franckowiak JD, Hoffman D, Skadsen R, Steffenson BJ (1993) A molecular, isozyme and morphological map of barley (Hordeum vulgare) genome. Theor Appl Genet 86:705–712
Konishi T, Linde-Laursen I (1988) Spontaneous chromosomal rearrangements in cultivated and wild barleys. Theor Appl Genet 75:237–243
Kosterin OE, Pukhnacheva NV, Gorel FL, Berdnikov VA (1999) Location of the breakpoints of four reciprocal translocations involving linkage group V and their influence on recombination distances between neighboring markers. Pisum Genet 31:13–20
Künzel G, Waugh R (2002) Integration of microsatellite markers into the translocation-based physical RFLP map of barley chromosome 3H. Theor Appl Genet 105:660–665
Künzel G, Korzun L, Meister A (2000) Cytologically integrated physical restriction fragment length polymorphism maps for the barley genome based on translocation breakpoints. Genetics 154:397–412
Lacasa-Benito I, Cuadrado A, Muñoz P, Moralejo M, Cistue L, Castillo A, Vallés P, Romagosa I (2005) Molecular and cytological study of the reciprocal translocation present in the drought stress resistant barley “Albacete”. Interdrought II (Poster), Rome, September 2005
Lehmensiek A, Eckermann PJ, Verbyla AP, Appels R, Sutherland MW, Daggard GE (2005) Curation of wheat maps to improve map accurary and QTL detection. Aust J Agric Res 56:1347–1354
Li JZ, Sjakste TG, Röder MS, Ganal MW (2003) Development and genetic mapping of 127 new microsatellite markers in barley. Theor Appl Genet 107:1021–1027
Liu Z-W, Biyashev RM, Saghai Maroof MA (1996) Development of simple sequence repeat DNA markers and their integration into a barley linkage map. Theor Appl Genet 93:869–876
Livingstone KD, Churchill G, Jahn MK (2000) Linkage mapping in populations with karyotypic rearrangements. J Hered 91:423–428
Mago R, Spielmeyer W, Lawrence GJ, Lagudah GJ, Ellis JG, Pryor A (2002) Identification and mapping of molecular markers linked to rust resistance genes located on chromosome 1RS of rye using wheat-rye translocation lines. Theor Appl Genet 104:1317–1324
Mahama AA, Palmer RG (2003) Translocation in soybean classical genetic linkage groups 6 and 8. Crop Sci 43:1602–1609
Mayer K, Taudien S, Martis M, Šimková H, Suchánková P, Gundlach H, Wicker T, Petzold A, Felder M, Steuernagel B, Scholz U, Graner A, Platzer M, Doležel J, Stein N (2009) Gene content and virtual gene order of barley chromosome 1H. Plant Physiol 151:496–505
Olson M, Hood L, Cantor C, Doststein D (1989) A common language for physical mapping of the human genome. Science 254:1434–1435
Payne RW, Murray DA, Harding SA, Baird DB, Soutar DM (2009) GenStat for windows (12th edition) introduction. VSN International, Hemel Hempstead
Pillen K, Binder A, Kreuzkam B, Ramsay L, Waugh R, Förster J, Leon J (2000) Mapping new EMBL-derived barley microsatellites and their use in differentiating German barley cultivars. Theor Appl Genet 101:652–660
Qi X, Stam P, Lindhout PL (1996) Comparison and integration of four barley genetic maps. Genome 39:379–394
Ramsay L, Macaulay M, degli Ivanissevich S, MacLean K, Cardle L, Fuller J, Edwards KJ, Tuvesson S, Morgante M, Massari A, Maestri E, Marmiroli N, Sjakste T, Ganal M, Powell W, Waugh R (2000) A simple sequence repeat-based linkage map of barley. Genetics 156:1997–2005
Sharma S, Bhat PR, Ehdaie B, Close TJ, Lukaszewski AJ, Waines JG (2009) Integrated genetic map and genetic analysis of a region associated with root traits on the short arm of rye chromosome 1 in bread wheat. Theor Appl Genet 119:783–793
Smilde W, Haluškove J, Sasaki T, Graner A (2001) New evidence for the synteny of rice chromosome 1 and barley chromosome 3H from rice expressed sequence tags. Genome 44:361–367
Sybenga J (1975) Meiotic configurations. Monographs on theoretical and applied genetics. Springer, Berlin
Tadmor Y, Zamir D, Ladizinsky G (1987) Genetic mapping of an ancient translocation in the genus Lens. Theor Appl Genet 73:883–892
Van Ooijen JW (2006) JoinMap® 4, software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Varshney RK, Marcel TC, Ramsay L, Russell J, Röder MS, Stein N, Waugh R, Langridge P, Niks RE, Graner A (2007) A high-density barley microsatellite consensus map with 775 SSR loci. Theor Appl Genet 114:1091–1103
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Waugh R, Bonar N, Baird E, Thomas B, Graner A, Hayes P, Powell W (1997) Homology of AFLP products in three mapping populations of barley. Mol Gen Genet 255:(31)1–321
Wenzl P, Carling J, Kudrna D, Jaccoud D, Huttner E, Kleinhofs A, Kilian A (2004) Diversity arrays technology (DArT) for whole-genome profiling of barley. Proc Natl Acad Sci USA 101:9915–9920
Wenzl P, Li H, Carling J, Zhou M, Raman H, Paul E, Hearnden P, Maier C, Xia L, Caig V, Ovesná J, Cakir M, Poulsen D, Wang J, Raman R, Smith KP, Muehlbauer GJ, Chalmers KJ, Kleinhofs A, Huttner E, Kilian A (2006) A high-density consensus map of barley linking DArT markers to SSR, RFLP and STS loci and agricultural traits. BMC Genomics 7:206
Acknowledgments
This study has been supported by the Spanish Ministry of Science and Innovation (Project AGL2008-05541-C02 and the Agrogenomics CONSOLIDER). We are grateful to Dr. Martin Boer who provided us with simulated data which helped us in better understanding the mapping problems arising from the presence of a reciprocal translocation. Comments by the editor and two referees are gratefully acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Hackett.
Appendix
Appendix
Cytogenetical background
During meiotic prophase I homologous chromosomes interact with each other through precise synapsis and crossover recombination. In the case of a reciprocal translocation heterozygote, saturated pairing gives rise to a quadrivalent (association of 4 chromosomes) involving the two translocation chromosomes and their non-translocation counterparts (Burnham 1962; Sybenga 1975). Various configurations can be observed at diakinesis–metaphase I, depending on the number and positions of the crossing overs: zig-zag shapes, rings and open forms of 2–4 chromosomes, and univalents. Crossing overs may occur in three types of chromosomal segments: the interchanged segment, the interstitial segment (between translocation points and centromeres) and the non-interchanged segments (chromosome arms distal to the centromeres).
With a single crossing over in the distal parts of the pairing segments, the viability of the resulting gametes depends on the co-orientation of the centromeres at metaphase I. This may lead to alternate segregation (corresponding with a zig-zag configuration of the quadrivalent) or adjacent segregation (corresponding with a ring configuration of the quadrivalent). For adjacent segregation it is possible to distinguish between two types: in adjacent-2 segregation homologous centromeres end up in the same telophase I cell, in adjacent-1 segregation they end up in different telophase I cells. Adjacent segregation leads to gametes with a translocation and a non-translocation chromosome, containing a duplicated and a deleted segment; this leads to non-viable gametes. Alternate segregation occurs when the chromosomes on opposite sides of the translocation cross-segregate into the same pole. In the latter case, four balanced gametes are formed: 50% carry the normal chromosome arrangement, and 50% carry translocated chromosomes. Hence, depending on the relative frequency of alternate segregation, individuals heterozygous with regard to a translocation will produce a fraction of non-viable gametes. This results in a reduction of fertility; it is called semi-sterility.
If a single crossing over occurs in one of the interstitial segments followed by alternate or adjacent-1 segregation, 50% of the gametes produced (the non-recombinants) are of the parental type, and the other 50% (recombinants) carry a segment deficiency and a duplication and are non-viable. The same crossing over followed by adjacent-2 segregation produces only non-viable gametes. As a consequence, crossing overs in one of the interstitial segments leads to a reduction of recombination in the interstitial segments. It should be noticed that in barley, alternate segregation occurs more frequently than the other segregation types.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Farré, A., Lacasa Benito, I., Cistué, L. et al. Linkage map construction involving a reciprocal translocation. Theor Appl Genet 122, 1029–1037 (2011). https://doi.org/10.1007/s00122-010-1507-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1507-2