
Abstract
The damage caused by the parasitic root cyst nematode Globodera pallida is a major yield-limiting factor in potato cultivation . Breeding for resistance is facilitated by the PCR-based marker ‘HC’, which is diagnostic for an allele conferring high resistance against G. pallida pathotype Pa2/3 that has been introgressed from the wild potato species Solanum vernei into the Solanum tuberosum tetraploid breeding pool. The major quantitative trait locus (QTL) controlling this nematode resistance maps on potato chromosome V in a hot spot for resistance to various pathogens including nematodes and the oomycete Phytophthora infestans. An unstructured sample of 79 tetraploid, highly heterozygous varieties and breeding clones was selected based on presence (41 genotypes) or absence (38 genotypes) of the HC marker. Testing the clones for resistance to G. pallida confirmed the diagnostic power of the HC marker. The 79 individuals were genotyped for 100 single nucleotide polymorphisms (SNPs) at 10 loci distributed over 38 cM on chromosome V. Forty-five SNPs at six loci spanning 2 cM in the interval between markers GP21-GP179 were associated with resistance to G. pallida. Based on linkage disequilibrium (LD) between SNP markers, six LD groups comprising between 2 and 18 SNPs were identified. The LD groups indicated the existence of multiple alleles at a single resistance locus or at several, physically linked resistance loci. LD group C comprising 18 SNPs corresponded to the ‘HC’ marker. LD group E included 16 SNPs and showed an association peak, which positioned one nematode resistance locus physically close to the R1 gene family.
Similar content being viewed by others

Introduction
The damage caused by the parasitic root cyst nematode Globodera pallida (Greco 1988; Mugniéry and Phillips 2007) is a major yield-limiting factor in potato cultivation. Under poor nematode management, yield losses induced by G. pallida can range from 20 to 70%. The cultivation of nematode-resistant varieties is a solution to this problem. The introgression and maintenance of resistance against G. pallida in the breeding pool is therefore an important goal in the development of new potato varieties. Due to its quantitative nature, the phenotypic evaluation of resistance to G. pallida is time consuming, costly and often ambiguous. DNA markers could facilitate breeding for resistance, when they are diagnostic for favourable alleles and thus allow for a precise and efficient selection of resistant clones at an early stage in the selection process.
Molecular mapping has repeatedly revealed a major quantitative resistance locus (QRL) against G. pallida on potato chromosome V (Kreike et al. 1994; Rouppe van der Voort et al. 1998, 2000; Caromel et al. 2003; Sattarzadeh et al. 2006). This QRL is tagged by the DNA markers GP21 and GP179 flanking a genetic interval of 3 cM (Meksem et al. 1995). This region of the potato genome is known as a hot spot for qualitative and quantitative resistance factors to various pathogens (reviewed in Gebhardt and Valkonen 2001). One of the resistance factors in the GP21—GP179 interval is the R1 gene for resistance to Phytophthora infestans causing late blight, which is a member of a clustered gene family having a CC (coiled coil), a NB (nucleotide binding) and a LRR (leucine rich repeat) domain (Ballvora et al. 2007). Physically close to the R1 locus, in fact, within one of the R1 family members, maps the PCR-based marker ‘HC’. The HC marker was developed based on SNPs (single nucleotide polymorphisms) linked to the G. pallida QRL and was shown to be diagnostic for high resistance to G. pallida pathotypes Pa2/3 in modern potato varieties (Sattarzadeh et al. 2006). The strong association of the HC marker with resistance to G. pallida may result from HC being located in the resistance gene itself, or, more likely, HC being physically separated from, but in strong linkage disequilibrium (LD) with the resistance gene(s). In the latter case, HC is part of a haplotype block of unknown size.
Molecular identification of the gene(s) underlying the G. pallida QRL requires increased genetic resolution, which can be achieved by generating a large segregating population and analysing it for rare recombination events flanking the target locus. Alternatively, historical recombination events in contemporary germplasm may be analysed, which have occurred during the meiotic cycles since the resistance was introgressed into modern potato varieties from the wild, tuber-bearing species Solanum vernei (Sattarzadeh et al. 2006). In the absence of population substructure, the estimator for the genetic distance between a marker and the targeted resistance locus is given by the amount of linkage disequilibrium (LD), that is, the non-random association of alleles at two or more loci (Tenesa et al. 2003). In populations of individuals related by descent, marker loci that are in LD with each other may show a gradient of the strength of their association with a QTL, thereby permitting QTL fine mapping. SNPs, which are found with high frequency in amplicons generated from genomic DNA of heterozygous, diploid and tetraploid individuals (Rickert et al. 2003; Simko et al. 2006) are most informative about LD analysis in potato. Moreover, SNPs allow estimating the allele dosage in tetraploid individuals (Rickert et al. 2003). Haplotypes, which are particular combinations of alleles at physically linked loci, can be more informative than individual SNPs. In potato, the inference of haplotypes from sequence data is complicated because SNP data are unphased due to the heterozygocity of individual genotypes.
In this paper, we describe the results of genotyping an unstructured population of tetraploid varieties and breeding clones with and without the HC marker for 100 SNPs at ten loci on potato chromosome V. The aim of our study was (1) to estimate the size of the haplotype block around the HC marker and (2) to test whether LD analysis reveals more precisely the position of the gene(s) underlying the major QRL to G. pallida on chromosome V.
Materials and methods
Plant material
Eighteen varieties and 189 tetraploid clones from breeding programs were provided by Saka-Ragis Pflanzenzucht (SaRa), Böhm-Nordkartoffel Agrarproduktion (BNA), Bavaria Saat (by courtesy of A. von Zwehl), HZPC Holland B.V., Averis Seeds B.V., Karna, E.J. Feunekes, Kweekbedrijf Prummerl and B.F. Leestemaker & A. Smid (Table 1). The clones from Saka-Ragis Pflanzenzucht and Böhm-Nordkartoffel Agrarproduktion are identified with ‘S’ and ‘B’, respectively, followed by a running number from 1 to 120. The S and B clones were sampled from the two companies conducting breeding programs for resistance to G. pallida and late blight, excluding full sib clones. The clones represented a broad range of breeding materials.
Assessment of resistance to G. pallida
Pot inoculation tests were carried out according to Kort et al. (1977). The resistance phenotype was quantified as the average number of newly formed cysts per pot. The BNA clones were tested once with two to three replicates per genotype at the ‘Landwirtschaftskammer Niedersachsen’ (Hannover, Germany) using for inoculation the G. pallida population ‘Kalle’ (pathotype Pa2). The SaRa clones were evaluated in 3 years (2004, 2005 and 2006) with four replicates per genotype at the ‘Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern’ (Rostock, Germany). Here the ‘Chavornay’ population (pathotype Pa2/3) was used for inoculation. The G. pallida resistance phenotype of the varieties was obtained from the variety lists of the Netherlands (81e rassenlijst Landbouwgewassen, 2006, ISSN 0168-7484) and Germany (Beschreibende Sortenliste Kartoffeln, 2005, ISSN 1430-9777).
DNA isolation
Young, healthy potato leaves were harvested from each individual, freeze dried and stored in air-tight containers at −20°. Total genomic DNA was extracted from 0.3 to 0.4 g freeze dried leaf material according to Bormann et al. (2004).
SNP (single nucleotide polymorphism) genotyping
Amplicons were generated from 50 ng genomic DNA as template in 20 μl buffer (20 mM Tris–HCl, pH 8.4, 2.5 mM MgCl2, 50 mM KCl), including 0.25 μM of each primer (Table 2), 0.2 mM dNTP and 1.0 unit Taq DNA polymerase (Invitrogen Life Technologies, Freiburg, Germany). Standard cycling conditions were 3 min initial denaturation at 94°, followed by 39 cycles of 1 min denaturation at 94°, 1 min annealing at the appropriate T m (Table 2) and 1 min extension at 72°. Reactions were finished by 8 min incubation at 72°. PCR products were examined for quality on ethidium bromide stained agarose gels. Prior to sequencing, PCR products were purified with ExoSAP-IT® (USB Corporation, Cleveland, USA) according to supplier’s instructions. Amplicons were custom sequenced at the MPIZ DNA core facility using an Applied Biosystems Abi Prism 3730 sequencer (Weiterstadt, Germany) and BigDye-terminator v3.1 chemistry. Premixed reagents were from Applied Biosystems. Oligonucleotides were purchased from Invitrogen, Life Technologies (Freiburg, Germany) or QIAGEN GmbH (Hilden, Germany). Sequencing data of all individuals were aligned using the multalin interface page (Corpet 1988) to detect SNP positions. For estimating the dosage of SNP alleles in heterozygous, tetraploid individuals (3:1, 2:2 or 1:3), the sequence tracefiles were subjected to the Data Acquisition and Analysis Software (DAx) (http://www.dax.nl/daxapp1.htm).
SSR (simple sequence repeat) genotyping
A set of 19 SSR marker loci with a random genome wide distribution were genotyped to be used in the analysis of population substructure. The following 19 SSR-marker loci were analysed: StI004, StI013, StI020, StI022, StI023, StI024, StI028, StI043, StI047 and StI058 according to Feingold et al. 2005; STM0037, STM0038, STM1043, STM1052, STM1097, STM1104, STM1106, STM2012 and STM3012 according to Milbourne et al. 1998. SSR alleles were size separated on SPREADEX gels (Elchrom Scientific, Cham, Switzerland) using the SEA 2000 Electrophoresis System (Elchrom Scientific) according to manufacturer’s instructions. Gels were stained after electrophoresis with 10% SYBR™ Gold in 30 mM TAE buffer for 45 min and de-stained for 30 min in destaining Solution (Elchrom Scientific) in 12 mM TAE. Two to seven alleles were scored per SSR locus. Allele dosage (quadruplex, triplex, duplex, simplex or nulliplex) was estimated based on the number of alleles per individual and the band intensity of the alleles present in one individual relative to each other.
Pyrosequencing
The marker GP179 was analysed by pyrosequencing (Biotage AB, Uppsala, Sweden) according to the manufacturer’s instructions. A fragment of 186 base pairs was amplified using the primer combination GP179r-biotin and GP179f (Table 2). PCR was carried out in a total volume of 25 μl containing 4 mM Tris–HCl, pH 8.3, 20 mM KCl, 3 mM MgCl2, 0.5 μM each primer, 0.2 mM dNTPs, 1 unit Taq DNA Polymerase (Invitrogen Life Technologies, Freiburg, Germany). After initial denaturation at 94° for 2 min, 50 cycles of amplification were carried out starting at 93° for 45 s, followed by 45 s at 57° and 1 min at 72°, with a final extension at 75° for 10 min. PCR products were quantified on 1.5% agarose gels relative DNA standards. The pyrosequencing reactions were performed automatically with a PSQ 96 system (Biotage AB, Uppsala, Sweden) using a SNP reagent kit according to the manufacturer’s instructions and the sequencing primer GP179s (Table 2). All oligonucleotides were custom synthesized and HPLC-purified by SIGMA-Genosys, Germany.
Data analysis
Population structure was evaluated by the program STRUCTURE version 2.1 (Pritchard et al. 2000) using the genetic information provided by 113 SSR alleles at 19 loci with a genome wide distribution. The number k of underlying subpopulations was set to vary between two and ten. For each k value, two runs of 600,000 iterations were performed, of which the first 100,000 were discarded as burn in. An admixture model with independent allele frequencies was assumed, without a priori population information.
The degree of linkage disequilibrium (LD) between pairs of SNP markers was based on a statistical test of independence, assuming no population substructure. In tetraploid individuals, each bi-allelic SNP marker occurs in one of five genotype classes: 1/1/1/1, 1/1/1/2, 1/1/2/2, 1/2/2/2 or 2/2/2/2, with 1 and 2 corresponding to alternative nucleotide alleles. The genotype classes for each SNP locus were first defined by the observed combination and dosage of nucleotides. Then, tests for independence of pairs of SNP markers were performed on the frequencies of genotype-class combinations, using the Exact Test for RxC tables, available in SAS Proc Freq (SAS Institute Inc. 2004). The p-values resulting from testing all SNP pairs were a posteriori corrected for multiple testing, according to the Two-Stage Linear Step-Up Procedure (Benjamini et al. 2005), and the values after the correction (q-values) used as a measure of the degree of independence, so that a lower q-value corresponds to larger evidence against independence of a pair of SNPs. A threshold of 0.01 was taken for the false discovery rate.
Associations between SNP markers and nematode resistance phenotype were analysed with Fisher’s exact test on 2 × 2 contingency tables using SPSS (SPSS 13 for Windows, SPSS Inc. Chicago, IL, USA).
Results
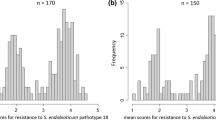
A total of 189 tetraploid-breeding clones were evaluated for presence of the HC marker. Thirty-one clones were HC positive. These and 10 HC positive varieties (Sattarzadeh et al. 2006) were selected as a sample population of 41 individuals having the HC marker. The control population negative for the HC marker consisted of 30 breeding clones and 8 varieties (Table 1). The status for resistance to G. pallida of the varieties was known (Table 1). When the ‘S’ clones were tested for resistance to G. pallida with the ‘Chavornay’ population (pathotype Pa2/3), all HC-positive clones were highly resistant (on average 0–6 newly formed cysts), whereas the HC-negative clones were susceptible (between 30 and 200 newly formed cysts), thus confirming the diagnostic power of the HC assay (Table 1). The HC-positive ‘B’ clones were therefore predicted to be resistant to G. pallida pathotype Pa2/3. When tested for resistance to G. pallida with population ‘Kalle’ (pathotype Pa2), the HC-positive ‘B’ clones were highly resistant (0–10 newly formed cysts) except clones B25 and B47, which were slightly susceptible (average cyst counts 15 and 29)(Table 1). The phenotypes of B25 and B47 may result from true recombination events or from errors in the resistance tests. The HC-negative ‘B’ clones were all susceptible (between 30 and 160 newly formed cysts) (Table 1).
Ten loci spanning a genetic distance of approximately 38 cM on chromosome V (Meksem et al.1995; Meyer et al. 2005; Niewöhner et al. 1995) were scored for SNPs in the 79 individuals (Table 2, Fig. 1, sequences in supplementary material). The loci ORF3, ORF12, ORF20, BA87d17t3 and BA213c14t7 were part of a contiguous genomic sequence of 417 kilobase pairs located in the 3 cM GP21-GP179 interval (Ballvora et al. 2007). The loci BA87d17t3 and BA213c14t7 are insertion ends of the BAC clones BA87d17 and BA213c14 and have been previously analysed in the QRL linkage study of Sattarzadeh et al. (2006). BA87d17t3 contains the two SNPs (BA87d17t3-SNP212, BA87d17t3-SNP444) used to design the HC marker (Sattarzadeh et al. 2006). The loci PRK and GP179 were part of a 41-kbp genomic sequence of BAC clone BA151m8 (gene bank accession AY730335). The loci StPto (potato homolog of the tomato Pto gene for resistance to Pseudomonas syringae (Martin et al. 1993, Leister et al. 1996) and 239E4left, tightly linked to the H1 gene for resistance to the root cyst nematode G. rostochiensis (Niewöhner et al. 1995, Bakker et al. 2004), were located outside the GP21-GP179 interval. In addition, 113 SSR alleles at 19 loci with a genome wide distribution were genotyped to assess population substructure. The likelihood of the data, conditional on the assumed number k of underlying subpopulations always increased with k, supporting the hypothesis of an unstructured population. Furthermore, in the model assuming population admixture we found that for k > 2 no individual was allocated to a single group; instead, individuals’ ancestry was represented as a mixture of all the main subgroups, so it would be impossible to partition individuals into groups.

Genetic and physical map of potato chromosome V. The ten loci analysed for SNPs are shown in bold print. The HC Marker was derived from 2 SNPs in the BA87d17t3 amplicon
One hundred SNPs, between four and 20 SNPs per amplicon, were scored and evaluated individually for association with the G. pallida QRL on chromosome V, by testing for unequal distribution of the SNP alleles between the HC-positive (resistant) sample population and the HC-negative (susceptible) control population. Forty-five SNPs were associated with resistance at P < 0.01 (Table 3). The significant SNPs were restricted to six loci, namely GP21, ORF3, ORF12, ORF20, BA87d17t3 and BA213c14t7 (Table 3, Fig. 2), which span a genetic distance of 2 cM.

Line diagram showing the P values for association between the SNPs and nematode resistance at the ten analysed loci. Y- axis: –Log P . X-axis: 100 SNPs in consecutive order. The loci are shown below the x-axis and are connected with the corresponding SNPs by broken lines. SNPs with P ≤ 0.01 (−Log P ≥ 2) were restricted to the loci GP21, ORF3, ORF12, ORF20, BA87d17t3 and BA213c14t7
Analysis of linkage disequilibrium between all pairs of SNP markers revealed LD among groups of SNPs, most strongly within the same amplicon. Figure 3 shows the LD matrix for the 100 SNPs. SNPs in LD to each other with q < 10−2 are shown as grey squares, whereas strong LD with q < 10−4 is represented by dark grey squares. Strong LD (q < 10−4) between SNPs in different amplicons was observed only in the 3-cM region between GP21 and GP179 and did not extend to StPto and 239E4left, located 5 and 35 cM, respectively, distal to GP179 (Figs. 1 and 3). The SNPs were then grouped using the criterion of strong LD (q < 10−4) between SNP pairs. The 45 SNPs associated with resistance sorted into six groups of 2–18 SNPs (Fig. 3, Table 4). Group C included 18 SNPs, which closely matched the distribution of the HC marker in the sample and control population. These 18 SNPs originated from loci GP21, ORF3, ORF20, BA87d17t3 and BA213c14t7. All SNPs in group C were associated with nematode resistance at P = 0.0001. Three SNPs (ORF3-SNP433, ORF3-SNP438, ORF3-SNP596) matched the distribution of the HC marker. Thirty-three of 41 individuals in the resistant sample population were heterozygous or homozygous for all 18 SNP alleles associated with resistance (Table 5), whereas the remaining eight individuals were homozygous for a portion of one to four of the 18 SNP alleles associated with susceptibility (Table 5). Three individuals of the susceptible control population carried exclusively SNP alleles associated with susceptibility, whereas the remaining 35 individuals were heterozygous for one to seven of the 18 SNP alleles associated with resistance. Inspection of the ‘recombinants’ in both the sample and control population (susceptible individuals heterozygous for some resistance SNP alleles and resistant individuals homozygous for some susceptibility SNP alleles) did not permit positioning more precisely the major QRL, for which the HC marker is diagnostic, within the 2 cm interval GP21 - BA213c14t7.

Linkage disequilibrium (LD) matrix for the SNPs. The 100 analysed SNPs are plotted on both axes. SNPs physically linked within the same amplicon are framed. LD significant with q ≤ 10−4 is indicated in dark grey and LD significant with 0.01 > q < 10−4 is indicated in light grey. The names of the ten loci analysed are shown on top of the matrix. The LD blocks C–G are shown on the vertical axes of the LD matrix, with the borders of LD blocks indicated by horizontal lines
SNP groups D1, D2, F and G were also not informative for fine mapping the QRL, D1 and D2 because all SNPs in these groups were similarly associated with resistance, F and G because each of these groups consisted only of SNPs within the same amplicon. However, LD group E including 16 SNPs at the loci GP21, ORF3, ORF20, BA87d17t3, BA213c14t7 and GP179 showed an association gradient, because only four SNPs at the loci BA87d17t3 and BA213c14t7 were associated with resistance at P < 0.01, whereas the remaining 12 SNPs in LD group E were not significantly associated with resistance. This peak of significant associations observed across six loci covering a genetic distance of 3 cM suggests that one or more genes for quantitative nematode resistance are located more closely to BA87d17t3 and BA213c14t7 than to GP21, ORF3 and ORF20 on the left, or GP179 on the right.
Discussion
A population of 79 tetraploid individuals was analysed in our study, which represents a maximum of 316 (4 × 79) chromosome V haplotypes. To capture 316 haplotypes, a minimum of nine bi-allelic SNP markers in linkage equilibrium is required (29 = 512). Genotyping 100 SNPs had therefore the potential to capture all chromosome V haplotypes that were actually present in the 79 individuals, at least the most common ones. The fact that most SNPs were in LD with one or more other SNPs also indicated that the SNP genotyping approached saturation. The 100 SNPs were distributed over ten loci. SNPs closely associated with resistance to G. pallida were restricted to the six loci distal to the marker locus GP179, spanning a genetic distance of 2 cM. The physical size of this region is around 1.7 Mb (Achenbach et al. manuscript in preparation). This region could be even larger, as no further marker was available for SNP analysis distal to GP21. The association analysis delimited therefore the genomic region harbouring the QRL at one side to the interval BA213c14t7–GP179, but not at the other.
Due to the heterozygocity and tetraploidy of the cultivated potato, the inference of specific haplotypes from SNP data is not straightforward. The SNP markers are unphased and the SNP allele dosage has to be considered in haplotype models (J. Neigenfind et al. 2008). Instead of haplotype models we used the strong LD observed among several SNPs to distinguish six LD groups, which included the SNPs associated with nematode resistance. The six groups discussed here represent only a subset of all LD groups present in the population, as LD groups comprising only SNPs not associated with nematode resistance were not considered and rare LD groups were not detected. Several different groups of SNPs in LD with each other were found, each group including SNPs associated with resistance to G. pallida. This indicates the existence of multiple alleles either at a single resistance locus or at several, physically closely linked loci, which control the quantitative resistance to G. pallida in this particular genomic region on potato chromosome V.
The most prominent resistance allele was associated with LD group C, which closely correlated with the HC marker. This is not surprising, as presence or absence of the HC marker was the criterion for selecting resistant and susceptible individuals in the population. No association gradient was observed within LD group C. The 18 SNPs in group C were all associated with resistance with similar strength and defined a large haplotype block comprising at least 2 cM or more than 400 kbp. Positioning the resistance gene(s) more precisely within this haplotype block was not possible based on the available SNP data. In the case of LD group C, two contrasting haplotype models could be deduced from the nucleotide alleles of the 18 SNP markers, one diagnostic of resistance similar to the HC marker, and the other one of susceptibility. The resistant haplotype was dominant. All heterozygous individuals having at least one dose of this resistant haplotype were resistant to G. pallida, whereas none of the susceptible individuals carried this haplotype. The gene(s) for resistance to G. pallida pathotype Pa2/3 associated with the HC marker, together with the SNP alleles in the resistant haplotype, have been introgressed into the European potato breeding pool (Solanum tuberosum) from the wild potato species Solanum vernei (Sattarzadeh et al. 2006). Large haplotype blocks may have been preserved due to limited recombination events in the breeding history of the germplasm analysed in our study. The pedigree information for G. pallida resistant varieties that is available from the ‘Potato Pedigree database’ (van Berloo et al. 2007, http://www.plantbreeding.wur.nl/potatopedigree/) is partial and contains ‘dead ends’. A number of S. vernei clones, which do not exist anymore, have been used several times in the pedigrees. The historical records do not allow identifying precisely the S. vernei resistance source and the number of meiotic generations, which separate contemporary germ plasm from the original introgressions. In general, variety pedigrees that can be traced back through the last 100 years include less than 10 meiotic generations (Love 1999; Gebhardt et al. 2004; van Berloo et al. 2007). Reduced recombination between homeologous chromosomes caused, for example, by inversions in the introgressed S. vernei segment relative to S. tuberosum, may also have contributed to the maintenance of a large haplotype block in the breeding populations. The existence of a 70-kbp inversion between S. demissum and S. tuberosum in this genome segment has been demonstrated (Ballvora et al. 2007). Suppression of recombination depending on the source and size of the introgression from wild species has been shown in tomato (Liharska et al. 1996; Kaloshian et al. 1998). Linkage disequilibrium extending over several centimorgan has also been observed by Simko et al. (2006) and Li et al. (2008) in different populations of tetraploid and diploid potato genotypes.
In contrast to LD group C, a peak of marker trait associations was observed within LD group E comprising 16 SNPs at 6 loci spanning 3 cM. Here, four SNPs at the loci BA87d17t3 and BA213c14t7 were associated with nematode resistance, whereas the SNPs at the flanking loci were not any more associated with nematode resistance. With the assumption that the QRL is controlled by one gene with multiple alleles, the association gradient observed in LD group E places this gene within or physically close to the genomic region delimited by BACs BA87d17 and BA213c14. This region harbours the R1 resistance gene and two paralogous members of the R1 gene family (Ballvora et al. 2007). When the alternative model is true, meaning that several genes in this genomic region control nematode resistance, at least one of these genes should be located within or physically close to the genomic region delimited by BACs BA87d17 and BA213c14. Association mapping in a large population of individuals lacking the almost qualitative HC marker would be of interest to better resolve the genetic architecture of the QRL based on weaker resistance alleles.
The individuals analysed in this study have been generated by inter-crossing tetraploid, highly heterozygous parents. LD decays more rapidly in outcrossing species when compared to selfing species, where recombination has no consequence for the genotype of homozygous individuals (Nordborg 2000). In potato, the diminishing effect of outcrossing on LD is counteracted by the vegetative propagation, which reduces the number of meiotic generations separating individual breeding clones and varieties (Gebhardt et al. 2004; van Berloo et al. 2007). The observed phenotypic and genotypic variability not only results from recombination, but also from a larger number, when compared with diploids, of chromosomal haplotype combinations that are possible in 4x by 4x crosses with tetrasomic inheritance. This might be one reason for the large genetic distances of similar size, which usually separate tetraploid individuals despite their relatedness by pedigree, and thereby the lack of population structure (Li et al. 2005). Another reason for lack of population structure might be an underlying uniformity of the genetic material due to its selective adaptation for general agronomic performance. Populations of varieties and breeding clones are highly suitable therefore to detect associations between markers and agronomic traits such as nematode resistance, due to the existence of large haplotype blocks and lack of population structure in this material. These properties make such populations less suitable for candidate gene verification due to lack of genetic resolution. Candidate genes for the QRL on potato chromosome V are, among other genes (Ballvora et al. 2007), the members of the R1 gene family. However, for identifying among all possible candidates the causal gene(s) for the observed nematode resistance phenotype in this genomic region, the full genome sequence of potato (http://www.potatogenome.net/) or the highly co-linear tomato (http://www.sgn.cornell.edu/about/tomato_sequencing.pl) will be needed, together with highly effective experimental systems for functional analysis of candidate genes.
References
Bakker E, Achenbach U, Bakker J, van Vliet J, Peleman J, Segers B, van der Heijden S, van der Linde P, Graveland R, Hutten R, van Eck H, Coppoolse E, van der Vossen E, Bakker J, Goverse A (2004) A high-resolution map of the H1 locus harbouring resistance to the potato cyst nematode Globodera rostochiensis. Theor Appl Genet 109:146–152
Ballvora A, Jöcker A, Viehöver P, Ishihara H, Paal J, Meksem K, Bruggmann R, Schoof H, Weisshaar B, Gebhardt C (2007) Comparative sequence analysis of Solanum and Arabidopsis in a hot spot for pathogen resistance on potato chromosome V reveals a patchwork of conserved and rapidly evolving genome segments. BMC Genom 8:112. doi:10.1186/1471-2164-8-112
Benjamini Y, Krieger A, Yekutieli D (2005) Two staged linear step up FDR controlling procedure. Unpublished report, available at http://www.math.tau.ac.il/~ybenja
Bormann CA, Rickert AM, Castillo Ruiz RA, Paal J, Lübeck J, Strahwald J, Buhr K, Gebhardt C (2004) Tagging quantitative trait loci for maturity-corrected late blight resistance in tetraploid potato with PCR-based candidate gene markers. Mol Plant Microbe Interact 17:1126–1138
Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890
Caromel B, Mugniery D, Lefebvre V, Andrzejewski S, Ellisseche D (2003) Mapping QTLs for resistance against Globodera pallida (stone) Pa2/3 in a diploid potato progeny originating from Solanum spegazzinii. Theor Appl Genet 106:1517–1523
Feingold S, Lloyd J, Norero N, Bonierbale M, Lorenzen J (2005) Mapping and characterization of new EST- derived microsatellites for potato (Solanum tuberolsum L.). Theor Appl Genet 111:456–466
Gebhardt C, Ballvora A, Walkemeier B, Oberhagemann P, Schüler K (2004) Assessing genetic potential in germplasm collections of crop plants by marker-trait association: a case study for potatoes with quantitative variation of resistance to late blight and maturity type. Mol Breed 13:93–102
Gebhardt C, Valkonen JPT (2001) Organization of genes controlling disease resistance in the potato genome. Annu Rev Phytopathol 39:79–102
Greco N (1988) Potato cyst nematodes: Globodera rostochiensis and G. pallida. Nematology circular no. 149. Florida Department of Agriculture and Consumer Services, Division of Plant Industry, Gainesville
Kaloshian I, Yaghoobi J, Liharska T, Hontelez J, Hanson D, Hogan P, Jesse T, Wijbrandi J, Simons G, Vos P, Zabel P, Williamson VM (1998) Genetic and physical localization of the root-knot nematode resistance locus Mi in tomato. Mol Gen Genet 257:376–385
Kort J, Ross H, Rumpenhorst HJ, Stone AR (1977) An international scheme for the identifying and classifying pathotypes of potato cyst- nematodes Globodera rostochiensis and G.pallida. Nematologica 23:333–339
Kreike CM, De Koning JRA, Vinke JH, Van Ooijen JW, Stiekema WJ (1994) Quantitatively-inherited resistance to Globodera pallida is dominated by one major locus in Solanum spegazzinii. Theor Appl Genet 88:764–769
Leister D, Ballvora A, Salamini F, Gebhardt C (1996) A PCR based approach for isolating pathogen resistance genes from potato with potential for wide application in plants. Nat Genet 14:421–429
Liharska TB, Koornneef M, van Wordragen M, van Kammen A, Zabel P (1996) Tomato chromosome 6: effect of the alien chromosomal segment on recombinant frequencies. Genome 39:485–990
Li L, Strahwald J, Hofferbert HR, Lübeck J, Tacke E, Junghans H, Wunder J, Gebhardt C (2005) DNA variation at the invertase locus invGE/GF is associated with tuber quality traits in populations of potato breeding clones. Genetics 170:813–821
Li L, Paulo M-J, Strahwald J, Lübeck J, Hofferbert H-R, Tacke E, Junghans H, Wunder J, Draffehn A, van Eeuwijk F, Gebhardt C (2008) Natural DNA variation at candidate loci is associated with potato chip color, tuber starch content, yield and starch yield. Theor Appl Genet 116:1167–1181
Love SL (1999) Founding clones, major contributing ancestors and exotic progenitors of prominent American potato cultivars. Am J Potato Res 76:263–272
Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW, Spivey R, Wu T, Earle ED, Tanksley SD (1993) Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262:1432–1435
Meksem K, Leister D, Peleman J, Zabeau M, Salamini F, Gebhardt C (1995) A high resolution map of the vicinity of the R1 locus on chromosome V of potato based on RFLP and AFLP markers. Mol Gen Genet 249:74–81
Meyer S, Nagel A, Gebhardt C (2005) PoMaMo: a comprehensive database for potato genome data. Nucleic Acids Res 33:D666–D670
Milbourne D, Meyer RC, Collins AJ, Ramsay LD, Gebhardt C, Waugh R (1998) Isolation, characterisation and mapping of simple sequence repeats. Mol Gen Genet 259:233–245
Mugniéry D, Phillips MS (2007) The nematode parasites of potato. In: Vreudgenhil D, Bradshaw J, Gebhardt C, Govers F, MacKerron DKL, Taylor MA, Ross HA (eds) Potato biology and biotechnology advances and perspectives. Elsevier, Amsterdam, pp 569–591
Neigenfind J, Gyetvai G, Basekow R, Diehl S, Achenbach U, Gebhardt C, Selbig J, Kersten B (2008) Haplotype inference from unphased SNP data in heterozygous polyploids based on SAT. BMC Genomics 9:356
Niewöhner J, Salamini F, Gebhardt C (1995) Development of PCR assays diagnostic for RFLP marker alleles closely linked to alleles Gro1 and H1, conferring resistance to the root cyst nematode Globodera rostochiensis in potato. Mol Breed 1:65–78
Nordborg M (2000) Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self- fertilization. Genetics 154:923–929
Pritchard JK, Stephens M, Donelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rickert AM, Kim JM, Meyer S, Nagel A, Ballvora A, Oefner PJ, Gebhardt C (2003) First-generation SNP/InDel markers tagging loci for pathogen resistance in the potato genome. Plant Biotechnol J 1:399–410
van der Rouppe Voort J, Lindeman W, Folkertsma R, Hutten R, Overmars H, van der Vossen E, Jacobsen E, Bakker J (1998) A QTL for broad-spectrum resistance to cyst nematode species (Globodera spp.) maps to a resistance gene cluster in potato. Theor Appl Genet 96:654–661
van der Rouppe Voort J, van der Vossen E, Bakker E, Overmars H, van Zandvoort P, Hutten R, Klein Lankhorst R, Bakker J (2000) Two additive QTLs conferring broad-spectrum resistance in potato to Globodera pallida are localized on resistance gene clusters. Theor Appl Genet 101:1222–1230
Sattarzadeh A, Achenbach U, Lübeck J, Strahwald J, Tacke E, Hofferbert HR, Rothsteyn T, Gebhardt C (2006) Single nucleotide polymorphism (SNP) genotyping as basis for developing a PCR-based marker highly diagnostic for potato varieties with high resistance to Globodera pallida pathotype Pa2/3. Mol Breed 18:301–312
Simko I, Haynes KG, Jones RW (2006) Assessment of linkage disequilibrium in potato genome with single nucleotide polymorphism markers. Genetics 173:2237–2245
Van Berloo R, Hutten RCB, van Eck HJ, Visser RGF (2007) An online potato pedigree database resource. Potato Res 50:45–57
Tenesa A, Knott SA, Carothers AD, Visscher PM (2003) Power of linkage disequilibrium mapping to detect a quantitative trait locus (QTL) in selected samples of unrelated individuals. Ann Hum Genet 67:557–566
Acknowledgments
This work was supported by grant 0313114A (GABI-CONQUEST project) of the German Federal Ministry for Education and Research (BMBF) in the framework of the GABI (genome analysis in the biological system plant) program.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. E. Bradshaw.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Achenbach, U., Paulo, J., Ilarionova, E. et al. Using SNP markers to dissect linkage disequilibrium at a major quantitative trait locus for resistance to the potato cyst nematode Globodera pallida on potato chromosome V. Theor Appl Genet 118, 619–629 (2009). https://doi.org/10.1007/s00122-008-0925-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-008-0925-x