
Abstract
This study characterises a series of 12 S-locus haplotype-specific F-box protein genes (SFB) in cherry (Prunus avium) that are likely candidates for the pollen component of gametophytic self-incompatibility in this species. Primers were designed to amplify 12 SFB alleles, including the introns present in the 5′ untranslated region; sequences representing the S-alleles S 1 , S 2 , S 3 , S 4 , S 4 ′, S 5 , S 6 , S 7 , S 10 , S 12 , S 13 and S 16 were cloned and characterized. [The nucleotide sequences reported in this paper have been submitted to the EMBL/GenBank database under the following accession numbers: PaSFB 1 (AY805048), PaSFB 2 (AY805049), PaSFB 3 (AY805057), PaSFB 4 (AY649872), PaSFB 4 ′ (AY649873), PaSFB 5 (AY805050), PaSFB 6 (AY805051), PaSFB 7 (AY805052), PaSFB 10 (AY805053), PaSFB 12 (AY805054), PaSFB 13 (AY805055), PaSFB 16 (AY805056).] Though the coding regions of six of these alleles have been reported previously, the intron sequence has previously been reported only for S 6 . Analysis of the introns revealed sequence and length polymorphisms. A novel, PCR-based method to genotype cultivars and wild accessions was developed which combines fluorescently labelled primers amplifying the intron of SFB with similar primers for the first intron of S-RNase alleles. Intron length polymorphisms were then ascertained using a semi-automated sequencer. The convenience and reliability of this method for the determination of the self-incompatibility (SI) genotype was demonstrated both in sweet cherry cultivars representing alleles S 1 to S 16 and in individuals from a wild population encompassing S-alleles S 17 to S 22 . This method will greatly expedite SI characterisation in sweet cherry and also facilitate large-scale studies of self-incompatibility in wild cherry and other Prunus populations.
Similar content being viewed by others

Introduction
The cherry (Prunus avium L.) is a diploid (2n=2x=16) member of the Rosaceae. The name ‘sweet cherry’ is generally used for commercial forms cultivated for their fruit, whereas ‘wild cherry’ relates to the ‘natural’ form of the species that is a frequent component of woodland margins and can be economically important as a timber tree. The species exhibits gametophytic self-incompatibility (GSI), controlled by the multi-allelic S-locus, in which incompatibility is determined by the haploid genotype of the pollen and the diploid genotype of the style (Crane and Lawrence 1929; McCubbin and Kao 2000). An understanding of the incompatibility genotype of commercial fruit cultivars and wild trees in natural stands or seed orchards is desirable to inform choices to maximize the set of fruit or seeds. Within wild cherry populations the SI status of an individual is of ecological importance for several reasons. Individuals are often widely scattered and thus the number and proximity of compatible pollen donors has a large impact on both seed-set and the genetic diversity of seedling populations. Wild cherry can propagate vegetatively by suckers and form large clonal groups (Ducci and Santi 1998), where all individuals contain the same S-alleles and are thus mutually incompatible. Such groups may dominate pollen donation to compatible neighbouring trees and have a significant impact upon the population genetics of the species. The methodologies currently available to determine S-genotype in cherry and other Prunus species are not well suited to large-scale population studies, and thus ecological and evolutionary processes in natural populations of these species have remained largely unstudied.
Gametophytic self-incompatibility is a mechanism adopted to prevent self-fertilisation and thus inbreeding in many plant species (de Nettancourt 2001; Franklin-Tong and Franklin 2003). The stylar component of GSI has been determined to be an S-RNase gene in the Scrophulariaceae (Xue et al. 1996), Solanaceae (Anderson et al. 1986; McClure et al. 1989) and Rosaceae (Bošković and Tobutt 1996; Sassa et al. 1996). S-RNases have been sequenced and characterised in all three families and two regions of high variability have been identified, HVa and HVb, with evidence of positive selection found for these regions (Ishimizu et al. 1998).
Within P. avium, six S-alleles (S 1 to S 6 ) were originally distinguished as a result of cross-pollination experiments and progeny testing carried out using sweet cherry cultivars (Crane and Brown 1937). A self-compatible, irradiation-induced variant of S 4 , called S 4 ′, has also been reported (Matthews 1970). These S-alleles have been correlated with stylar ribonucleases (Bošković and Tobutt 1996). Further S-RNases S 7 to S 16 were subsequently reported (Bošković and Tobutt 2001) and S 1 to S 16 have now been sequenced, though S 8 , S 11 and S 15 appear to be synonyms of S 3 , S 7 and S 5 , respectively (Sonneveld et al. 2001, 2003). Recently, a further six S-RNases S 17 to S 22 were identified in Belgian wild cherry populations (De Cuyper et al. 2005). Most of the alleles S 1 to S 16 have been observed in both wild cherry populations and cultivated sweet cherry cultivars. However, alleles S 10 and S 17 to S 22 appear, thus far, to be restricted to wild cherry populations (De Cuyper et al. 2005).
The allelic variants for Prunus S-RNase share several conserved regions and contain two introns, both of which exhibit considerable length polymorphism (Sonneveld et al. 2003; Tao et al. 1999). Techniques employing PCR and primers designed to anneal within conserved regions flanking both S-RNase introns have been employed to detect S-alleles in sweet cherry cultivars (Sonneveld et al. 2001, 2003; Tao et al. 1999; Wiersma et al. 2001) and also in populations of wild Prunus populations (De Cuyper et al. 2005; Kato and Mukai 2004). The primers of Sonneveld et al. (2003) were able to distinguish between alleles S 1 to S 22 by detecting the two S-RNase intron products, except in the case of S 13 where the first intron product was not amplified. The first intron amplification products range from 303 to 523 bp and the second intron amplification products range from 874 bp to 2.4 kbp. The first intron primers have recently been redesigned to amplify products under 500 bp to enable determination of size by means of fluorescently-labelled primers and a standard array on an ABI Prism 3100 semi-automated sequencer (Sonneveld et al. 2005b).
Whereas the stylar (S-RNase) component of the GSI mechanism has been extensively studied, the candidate pollen component genes for GSI in the Scrophulariaceae and Solanaceae have only recently been identified. Initial studies (Lai et al. 2002) in Antirrhinum (Scrophulariaceae) identified an F-box gene (AhSLF-S 2 ), specifically expressed in the pollen and tapetum, located ~9 kbp downstream of the S 2 S-RNase, which was thought to be a good candidate gene for the pollen component of GSI in this species. More recently Qiao et al. (2004b) have published supporting evidence that AhSLF-S 2 physically interacts with S-RNases. They also found that SLF interacts with ASK1 and CUL1-like proteins in Antirrhinum suggesting that they form multifactor SCF complexes recruiting substrates for degradation in pollen. Qiao et al. also provided evidence that inhibition of S-RNase activity occurs during compatible pollination via the 26S proteasome pathway. It is not yet clear if the allelic forms of AhSLF are sufficiently divergent to encode the pollen component of GSI in this species in its entirety. More recently transgenic experiments demonstrated that an S-locus F-box gene (PiSLF) identified in Petunia inflata (Solanaceae) was the pollen component gene for GSI in this species (Sijacic et al. 2004).
Recent efforts have also focused on the identification and characterisation of the pollen gene component of GSI within the Rosaceae. Initially Entani et al. (2003) and Ushijima et al. (2003) sequenced the S-loci of P. mume (Japanese apricot) and P. dulcis (almond), respectively. In P. mume, Entani et al. identified a candidate F-box gene named SLF, which exhibited S-haplotype specificity, was expressed exclusively in the pollen and was located together with the S-RNase gene, at the S-locus. Further F-box genes identified by Entani et al. which did not exhibit S-haplotype-specific polymorphism were designated SLFL. Ushijima et al. also identified an S-haplotype-specific F-box protein gene as the likely candidate for the pollen component of GSI in P. dulcis which they named SFB. S-haplotype-specific F-box (SFB) genes expressed specifically in the pollen were also identified in P. avium (Yamane et al. 2003a). The amino acid sequence identities between the P. mume SLF, P. dulcis SFB and P. avium SFB genes initially identified ranged from 79.9 to 87.9%. As such, the SLF/SFB genes from the three Prunus species may be considered homologous. Most recently, Romero et al. (2004) identified and characterised SFB gene homologues within the S-locus of P. armeniaca (apricot), which were also expressed specifically in the pollen.
When this study commenced, only the P. mume SLF and P. dulcis SFB genes had been reported. Of particular interest to the authors was the presence of an 85 bp intron in the 5′ untranslated region (5′UTR) reported for the P. dulcis SFB genomic clone S c . We speculated that if similar introns were associated with the P. avium SFB genes and exhibited S-haplotype-specific polymorphism; this might be exploited to elucidate the S-genotype in the same way that intron polymorphism in S-RNase alleles has proved useful. Subsequently, Yamane et al. (2003b) reported PaSFB 3 and PaSFB 6 from P. avium and noted the presence of an intron in the 5′UTR of PaSFB 6 . Recently Ikeda et al. (2004) have isolated and characterised the SFB genes associated with S 1 , S 2 , S 4 and S 5 from P. avium, but did not characterise the 5′UTR of these alleles.
The initial aim of this study was to isolate and characterise PaSFB alleles representing S-haplotypes S 1 to S 16 , to gain further understanding of the evolutionary relationships and phylogeny of SFB alleles within Prunus and also to investigate the potential for distinguishing S-alleles in wild populations and sweet cherry cultivars on the basis of possible intron length polymorphism associated with the 5′UTR of these genes. Subsequently, PCR was optimised to allow the length polymorphism of the SFB intron and the first intron of S-RNase to be amplified and scored simultaneously in a single reaction or ‘multiplex’.
Materials and methods
Plant material and genomic DNA extraction
To isolate P. avium SFB alleles for S 1 to S 16 , a range of cultivars with various combinations of S 1 to S 16, including the self-compatible allele S 4 ′, were utilised: ‘Skeena’ (S 1 S 4 ′), ‘Wildstar 1’ (S 1 S 14 ), ‘Victor’ (S 2 S 3 ), ‘Rodmersham Seedling’ (S 3 S 16 ), ‘Napoleon’ (S 3 S 4 ), ‘Sonata’ (S 3 S 4 ′), ‘Inge’ (S 4 S 9 ), ‘Late Black Bigarreau’ (S 4 S 5 ), ‘Governor Wood’ (S 3 S 6 ), Orleans-171 (S 7 S 10 ), ‘Burlat’ (S 3 S 9 ), ‘Flamentiner’ (S 5 S 12 ), ‘Goodnestone Black’ (S 5 S 13 ) and ‘Fice’ (S 3 S 14 ). The genotypes for most of these are reported in a compilation (Tobutt et al. 2004) and those of ‘Wildstar 1’ and ‘Fice’ were determined in preliminary work. A further selection of genotyped cherry cultivars representing each S-allele from S 1 to S 16 at least twice was employed for subsequent evaluation of the reliability of the method. As mentioned previously, S 8 , S 11 and S 15 are duplicates of S 3 , S 7 and S 5 , respectively, and are therefore not included in this study. Accessions from an English ancient woodland representing S 17 to S 22 were also included to allow some characterisation of intron length polymorphism of these other alleles. Two dormant buds of each cultivar were peeled, flash frozen and ground with two ball bearings in a RetschMM2000 mill. Genomic DNA (gDNA) was then extracted using a scaled down CTAB extraction (Doyle and Doyle 1987) incorporating the addition of 1% (v/v) β-mercaptoethanol and 2% (w/v) polyvinyl pyrollidine (PVP 40) to the extraction buffer.
Isolation of SFB homologues in P. avium
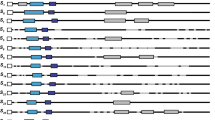
Sequence similarity analysis was carried out using four SFB alleles identified in P. dulcis (accessions: AB092966, AB092967, AB079776 and AB081648) (Ushijima et al. 2003) and three homologous SFB/SLF alleles identified in P. mume (accessions: AB092621, AB092622 and AB092645) (Entani et al. 2003) using the DNAstar Megalign (Lasergene) software. Degenerate primers, F-BOX5′A (TTK SCH ATT RYC AAC CKC AAA AG) and F-BOX3′A (WAT TGA GWA ARR SYA AAS TTT CTA), were designed to anneal within conserved regions identified upstream of a putative intron within the 5′UTR and towards the 3′ end of the coding sequence, respectively (Fig. 1).

Location of primers used during P. avium SFB gene cloning and PCR evaluation in relation to the 5′UTR intron (hatched region), start (ATG) and stop (*) codons
Amplification products representing genomic SFB clones for S-alleles S 1 to S 16 from the cultivars described previously were generated using the proof reading KOD DNA polymerase (Invitrogen) in PCR utilising 100 ng of gDNA as template in a 30 μl reaction mix (1×KOD buffer, 0.2 mM dNTP, 1 mM magnesium sulphate, 0.5 μM forward and reverse primers, 1 U KOD polymerase). PCR cycling conditions were 95°C for 2 min followed by 10 cycles of 94°C for 30 s, 60°C for 60 s with a reduction in annealing temperature of 1°C per cycle, 68°C for 90 s, then 25 cycles of 94°C for 30 s, 50°C for 60 s, 68°C for 90 s and a final cycle of 68°C for 10 min.
Amplification products were size fractionated by electrophoresis on 1.5% (w/v) agarose/1×TAE (40 mM tris-acetate, 10 mM EDTA) gels containing 0.5 mg/ml ethidium bromide and visualised by UV illumination. Amplification products of the expected size (~1.2 kbp) were excised using a clean scalpel. DNA was extracted and purified from the gel slices using the QIAEX II kit (Qiagen) following the manufacturer’s instructions exactly. Amplification products generated from each cultivar were subsequently cloned into the vector pCR4-TOPO (Invitrogen), transformed into TOP10 chemically competent cells (Invitrogen) and screened using colony PCR. Plasmid DNA was then prepared using a mini-spin kit (Qiagen) and inserts sequenced using M13 forward and reverse primers and an internal primer, F-BOX360 (AGA ATT TCA ATG GTC TCT TTT TTC C), designed specifically for this study.
Phylogenetic tree construction
Nucleotide sequence data for the 11 P. avium (PaSFB) alleles presented in this study were aligned using Clustal X (Thompson et al. 1997) with four SFB alleles from P. dulcis (PdSFBa: AB092966, PdSFBb: AB092967, PdSFBc: AB079776, PdSFBd: AB081648), three from P. mume (PmSFB 1 : AB101440, PmSFB 7: AB101441, PmSLF 9: AB092645) and three from P. armeniaca (ParmSFB 1 : AY587563, ParmSFB 2: AY587562, ParmSFB 4 : AY587564). Additionally, several non-S-haplotype-specific F-box protein genes were included in the alignment: five from P. mume (PmSLFL1-S 1 : AB0902623, PmSLFL1-S 7 : AB0902624, PmSLFL2-S 1 : AB0902625, PmSLFL2-S 7 : AB0902626, PmSLFL3-S 7 : AB0902627) and two from P. dulcis (PdSLFc: AB101659, PdSLFd: AB101660). Phylogenetic reconstruction was performed using parsimony in PAUP* (Swofford 2003) using 1,000 replicate heuristic searches with 100 random addition sequence replicates with TBR branch-swapping in effect. Support for the relationships inferred from the two most parsimonious trees recovered was measured by performing 1,000 heuristic bootstrap replicates (Felsenstein 1985) using PAUP*.
Development of novel methodology to determine S-genotype in P. avium
Fluorescently labelled primers (Applied Biosystems) were employed to amplify both the SFB intron and the S-RNase first intron of the full range of alleles S 1 to S 22 . Primer pair F-BOX5′A (6-FAM-TTK SCH ATT RYC AAC CKC AAA AG) and F-BOXintronR (CWG GTA GTC TTD SYA GGA TG) were designed to amplify the intron observed in the 5′UTR of the SFB alleles isolated (Fig. 1). These were combined in conjunction with consensus primers flanking the S-RNase first intron PaSPcons-F1 (VIC-MCT TGT TCT TGS TTT YGC TTT CTT C) and PaC1cons-R1 (GCC ATT GTT GCA CAA ATT GA) (Sonneveld et al. 2005b) in PCR. Reactions of 8 μl volume, containing approximately 1.25 ng P. avium gDNA, 1×Qiagen Multiplex PCR master mix (Qiagen) and 2 μM of each primer were used. Touch-down PCR was employed with cycling of 95°C for 15 min followed by 10 cycles of 94°C for 30 s, 55°C for 90 s with a reduction in temperatures of 0.5°C per cycle, 72°C for 60 s, then 25 cycles of 94°C for 30 s, 48°C for 90 s, 72°C for 60 s and a final cycle of 60°C for 30 min.
Following a 1:20 dilution, fluorescently tagged PCR amplification product sizes were determined using an ABI Prism 3100 semi-automated sequencer following the manufacturer’s instructions. Data for the SFB products were collected and allele sizes determined using GENESCAN and GENOTYPER software (Applied Biosystems), respectively. To corroborate further the scores generated for known S-haplotypes, data for the second intron of S-RNase were employed as previously described by Sonneveld et al. (2003).
Results
Isolation of SFB alleles from P. avium
For all but two alleles, the PCR strategy adopted here generated amplification products representing the intron associated with the 5′UTR and approximately 97% of the SFB gene coding region, with approximately 30 nt absent from the 3′ end. Although several attempts were made to isolate PaSFB 9 and PaSFB 14 both alleles proved recalcitrant to cloning, despite the use of a range of 3′ primers and several modifications to the PCR protocol described previously. However, P. avium SFB alleles were isolated successfully and characterised from various cultivars: PaSFB 1 from ‘Skeena’ (S 1 S 4 ′) and ‘Wildstar 1’ (S 1 S 14 ), PaSFB 2 from ‘Victor’ (S 2 S 3 ), PaSFB 3 from ‘Napoleon’ (S 3 S 4 ) and ‘Rodmersham Seedling’ (S 3 S 16 ), PaSFB 4 from ‘Napoleon’ (S 3 S 4 ) and ‘Inge’ (S 4 S 9 ), PaSFB 4 ′ from ‘Sonata’ (S 3 S 4 ′), PaSFB 5 from ‘Late Black Bigarreau’ (S 4 S 5 ), PaSFB 6 from ‘Governor Wood’ (S 3 S 6 ), PaSFB 7 and PaSFB 10 from Orleans-171 (S 7 S 10 ), PaSFB 12 from ‘Flamentiner’ (S 5 S 12 ), PaSFB 13 from ‘Goodnestone Black’ (S 5 S 13 ) and PaSFB 16 from ‘Rodmersham Seedling’ (S 3 S 16 ). In three cases where SFB alleles were isolated from more than one cultivar (i.e. S 1 , S 3 and S 4 ) identical sequence data were generated from both cultivars. If the same sequence was isolated from two or more cultivars possessing an S-allele in common then that sequence was considered to represent the shared allele. Subsequent additional sequences were attributed to the remaining allele. The significance of a 4 bp deletion distinguishing the coding sequence of PaSFB 4 ′ from PaSFB 4 has previously been reported (Sonneveld et al. 2005a; Ushijima et al. 2004).
Characterisation of P. avium SFB gene coding regions
Pairwise comparisons at the nucleotide level revealed lowest and highest degrees of sequence similarity of 75.9 and 90.3%, respectively, between the PaSFB gene coding regions isolated and the published P. dulcis and P. mume sequences used in the initial analysis. Further comparison with the published P. avium SFB genes for haplotypes S 1 to S 6 revealed sequence similarities between 99.5 and 100% for the coding region of these genes at the nucleotide level. As the sequences previously reported were isolated from cultivars different from those used in this study, the homologies provide further confirmation that the polymorphism observed between PaSFB genes of different S-haplotypes is generally consistent between individuals.
However, for PaSFB 1 eight points of conflict were observed between the nucleotide sequences reported by Ikeda et al. (2004) for ‘Seneca’ (S 1 S 5 ) (AB111518) and that reported here. These discrepancies result in a difference of eight amino acids and one missing residue in the predicted protein sequence for this gene. The sequence for PaSFB 1 isolated during this study was found to be identical in two cultivars of very different origin, ‘Wildstar 1’ (S 1 S 14 ), a wild accession selected for timber production, and ‘Skeena’ (S 1 S 4 ′), a modern fruit cultivar. Several plasmid preparations for PaSFB 1 were sequenced from these cultivars and robust sequence data were generated giving a high degree of confidence in the results reported here. In PaSFB 2 , two nucleotides differed between the coding sequence reported here for the cultivar ‘Victor’ and that reported by Ikeda et al. (2004), for ‘New York 54’ (S 2 S 6 ) (AB111519). The first nucleotide conflict within the two sequences (G:A at position 544) results in an amino acid substitution (threonine:alanine) between the two sequences. The second nucleotide conflict (Y:T at position 555) represents a degeneracy in the previously reported sequence but does not result in an amino acid substitution. For the purpose of this report the sequence data for PaSFB 1 and PaSFB 2 generated in this study were used for all sequence comparisons.
The predicted amino acid sequence for the P. avium SFB alleles isolated in this study revealed the presence of both the F-box motif and two highly variable non-conserved regions, HVa and HVb (Fig. 2), which were previously reported (Ikeda et al. 2004; Ushijima et al. 2003) as common to Prunus SFB genes. Pairwise comparisons at the protein level revealed lowest and highest homologies of 73.2 and 90.9%, respectively, among the PaSFB coding regions isolated. Pairwise comparisons at the protein level between the PaSFB coding regions of the P. dulcis (SFB), P. mume (SLF/SFB) and P. armeniaca (SFB) sequences revealed homologies of 75.9–91.3%.

Comparison of amino acid sequences predicted from the coding regions of P. avium SFB alleles. Sequence analysis was carried out using the Clustal W method. Shading denotes conserved regions, dashes represent gaps. F-box and hypervariable regions HVa and HVb (as defined by Ikeda et al. 2004) are boxed
Phylogenetic comparison
Construction of a phylogenetic tree revealed two most parsimonious trees with 100% bootstrap support for the division between the SFB clade and the SLF/SLFL clade (Fig. 3). All of the F-box genes isolated in this study were placed, as expected, within the SFB clade. Note that the P. mume allele SLF 9 represents a true SFB homologue (Entani et al. 2003); the variable nomenclature of the S-locus F-box genes between species sometimes causes confusion. Well-supported division was also noted within the SFB clade with PdSFBa, PaSFB 4/4 ′ and ParmSFB 2 being distinct from the main SFB group. Other than the strong relationship expected between PaSFB 4 and PaSFB 4 ′ the most strongly supported relationships between SFB alleles were observed between PaSFB 6 and PaSFB 16 , PaSFB 2 and ParmSFB 1 and PaSFB 10 and PaSFB 13 , . Allelic forms of SFB often showed a closer relationship to those of other species than to others of their own species. This is similar to patterns observed in S-RNase phylogenies (data not shown) indicative of trans-specific evolution within Prunus. However, little similarity exists between the phylogenetic tree derived from the deduced amino acids of the SFB alleles presented here and that of the corresponding S-RNases (data not shown).

Phylogenetic analysis of Prunus F-box alleles showing the strict consensus for two most parsimonious trees (consistency index=0.634; retention index=0.727; tree length=3,340) constructed using Prunus F-box nucleotide sequence data for 11 P. avium (PaSFB) alleles presented in this study aligned with four SFB alleles from P. dulcis (PdSFBa: AB092966, PdSFBb: AB092967, PdSFBc: AB079776, PdSFBd: AB081648), three from P. mume (PmSFB 1 : AB101440, PmSFB 7: AB101441, PmSLF 9: AB092645) and three from P. armeniaca (ParmSFB 1 : AY587563, ParmSFB 2: AY587562, ParmSFB 4 : AY587564). Additionally, several non-S-haplotype-specific F-box protein genes were included in the alignment; five from P. mume (PmSLFL1-S 1 : AB0902623, PmSLFL1-S 7 : AB0902624, PmSLFL2-S 1 : AB0902625, PmSLFL2-S 7 : AB0902626, PmSLFL3-S 7 : AB0902627) and two from P. dulcis (PdSLFc: AB101659, PdSLFd: AB101660). Phylogenetic reconstruction was performed by means of parsimony in PAUP* using heuristic searches. Significant bootstrap values are given above the branches and are based on 1,000 heuristic replicates
Characterisation of introns present in the 5′UTR of allelic variants of PaSFB
A putative intron was found to be present in the 5′ UTR of all 11 P. avium SFB alleles isolated. Further examination of these intron regions revealed length and sequence polymorphisms (Fig. 4). Intron lengths ranged from 81 to 122 bp, with nucleotide sequence homologies ranging from 27.1 to 67%. A conserved motif (TGAG) was noted at the 5′ intron border in all alleles except for PaSFB 16 . A further conserved motif (TDCAG) at the 3′ intron border was observed in all alleles. The majority of the intron sequences contained 1–2 direct repeats of 6–10 bp at varying positions as well as a highly conserved HTAACTY motif towards the 3′ end of the intron. The exception to this was the short intron of PaSFB 16 in which no direct repeats were observed. A phylogenetic analysis of the SFB intron regions revealed little phylogenetic structure and the resultant tree showed little congruity to that derived from analysis of the SFB coding regions (data not shown).

Alignment of nucleotide sequences representing the intron regions associated with the 5′UTR of 11 P. avium SFB alleles. Sequences were aligned using Clustal W. Conserved regions are highlighted with conserved motifs (identical in 9 of the 11 alleles presented) underlined. Intron sequences correspond to the GenBank accession numbers: PaSFB 1 (AY805048), PaSFB 2 (AY805049), PaSFB 3 (AY805057), PaSFB 4 (AY649872), PaSFB 5 (AY805050), PaSFB 6 (AY805051), PaSFB 7 (AY805052), PaSFB 10 (AY805053), PaSFB 12 (AY805054), PaSFB 13 (AY805055), PaSFB 16 (AY805056)
Although of different compositions, the intron sequences of PaSFB 1 and PaSFB 4 were of equal length. The intron of the PaSFB 4 ′ allele, which contains a mutated form of the SFB 4 gene, was identical to that of PaSFB 4 in both length and composition. However, none of the other introns sequenced were of exactly the same length which indicated that this polymorphism might be exploited to develop a rapid screening method to determine S-genotype in Prunus.
Evaluation of length polymorphism of the PaSFB 5′UTR intron in combination with that of the S-RNase first intron enables rapid determination of S-genotype
Fluorescently labelled PCR primers were used in a multiplex reaction to amplify the putative intron in the SFB 5′UTR together with the S-RNase first intron. The majority of accessions amplified two products for each gene and similar amplification products were reliably generated from several individuals of each S-haplotype. SFB 5′UTR intron amplification products for alleles S 1 to S 22 generated electrophorograms displaying profiles consistent with a diploid harbouring two allelic forms of the same gene. By comparing amplification patterns of known genotypes, particular products could be correlated with particular alleles. Furthermore, for alleles S 1 to S 16 , SFB intron PCR amplification products were consistently in accordance with the sizes (178–202 bp) expected from the sequence data for the S 1 to S 16 SFB intron regions. This analysis revealed that not all alleles were distinguishable from each other (Table 1): SFB 1 , SFB 4 , SFB 9 and SFB 19 all generate products of 189 bp; SFB 5 and SFB 17 both generate products of 190 bp; SFB 10 and SFB 22 both generate products of 175 bp; and SFB 13 and SFB 14 both generate products of 191 bp. However, in each case these alleles are distinguishable from each other using the S-RNase first intron amplification product alone. Although the amplification products representing the SFB 9 and SFB 14 intron products were initially determined to be 189 and 191 bp, respectively, subsequent screens using a larger number of samples revealed amplification of these alleles to be unreliable. Amplification products were reliably generated for the S-RNase first introns for all alleles from S 1 to S 16 in accordance with Sonneveld et al. (2005b).
The majority of the S-haplotypes from S 1 to S 16 may be distinguished using size data generated from the S-RNase first intron amplification products, the notable exceptions being S 2 , S 7 and S 12 all of which generate an S-RNase first intron product of approximately 345 bp. These alleles occur frequently in wild cherry populations (De Cuyper et al. 2005) and distinguishing between them using traditional methods greatly increases the experimental inputs required to undertake population studies in this species. Fortunately, for each of these haplotypes, the amplification products of the SFB 5′UTR intron region differ in size, 187, 181 and 185 bp, respectively, for S 2 , S 7 and S 12 (Fig. 5). In general, the SFB 5′UTR intron product sizes generated for each haplotype serve to confirm the haplotype implied by the S-RNase first intron product. In the case of S 2 , S 7 and S 12 the SFB 5′UTR intron may be used to distinguish between haplotypes in a single PCR reaction, eliminating the need for subsequent analysis using agarose gel electrophoresis of the S-RNase second intron or S-allele-specific PCR amplification products.

Electrophorograms displaying fluorescently labelled (VIC) S-RNase first intron PCR amplification products generated for P. avium genotypes S 7 S 10 , S 2 S 14 , S 3 S 12 (a) and fluorescently labelled (6-FAM) SFB intron products for the same genotypes (b). Both S-alleles are indicated for each cultivar, as are relative peak intensities and amplification product sizes as estimated against an internal (LIZ500) size standard. Amplification products are labelled for the right hand peak and the left hand (PCR artefact) ignored. In addition, note that S 7 , S 2 and S 12 are indistinguishable with S-RNase primers but are easily distinguished with SFB intron primers
Discussion
Characterisation of allelic forms of P. avium SFB
Here we report the successful isolation of all P. avium SFB alleles from S 1 to S 16 with the exception of PaSFB 9 and PaSFB 14 . Several different primer combinations, designed to accommodate degeneracy within the available Prunus SFB sequences towards the 5′ and 3′ end of the gene, failed to generate amplification products representing either PaSFB 9 or PaSFB 14. This suggests that these alleles may deviate significantly from the amplified alleles at the primer sites chosen or that the PCR is negatively affected by the surrounding DNA conformation at these loci. Although SFB 5′UTR intron products were initially amplified for these two alleles this was not reproducible, again indicating that the primers designed may not be sufficiently robust to routinely amplify these alleles. However, both alleles are readily identifiable using S-RNase first intron products.
It has been postulated that S-RNase-mediated GSI evolved only once in the eudicots (Igic and Kohn 2001; Steinbachs and Holsinger 2002) and previous studies have also indicated that the S-RNases from the Scrophulariaceae, Solanaceae, and Rosaceae form a monophyletic clade (Igic and Kohn 2001; Xue et al. 1996). Our analyses revealed evidence both of trans-specific evolution and a level of diversity comparable to that observed between the S-RNases for the same 11 SFB alleles in P. avium. Two highly variable non-conserved regions, HVa and HVb containing sites under positive selection, were identified within the PaSFB amino acid sequences isolated by Ikeda et al. (2004). In the five novel P. avium SFB alleles reported here both variable regions were present and distinct from those found in previously reported SFB alleles, lending further support to the hypothesis that these variable regions may function to provide S-allele specificity within the GSI reaction.
Characterisation of P. avium SFB 5′UTR introns and their application in determining S-genotypes
The intron lengths associated with the 5′UTR of the PaSFB alleles isolated display polymorphism across sequence lengths of 81–122 bp. Compared to first and second intron lengths observed within Prunus S-RNase genes, this represents a relatively small size range. There may well be genetic factors constraining the SFB intron length imposed by their location in the 5′UTR, where longer insertions could inhibit translational efficiency. The presence of common elements in all the SFB introns examined, and repetitive elements within the longer introns, likely reflect sequence duplications and rearrangements that have occurred as the SFB gene family has evolved.
Until recently the most rapid method to characterise S-genotypes in Prunus has utilised PCR and agarose gel electrophoresis to characterise both first and second intron lengths. However, this method is relatively time consuming and unsuitable for the analysis of large numbers of individuals. De Cuyper et al. (2005) utilised S-RNase second intron consensus primers in their study of self-incompatibility genotypes in Belgian wild cherries. This study necessitated 65 second intron consensus PCRs, 136 allele-specific PCRs and 19 first intron consensus PCRs (220 total) and their subsequent agarose gel electrophoresis to obtain the S-genotype for 65 individuals. Recent redesign of S-RNase first intron primers to enable product lengths to be determined on a semi-automated sequencer (Sonneveld et al. 2005b) represents a significant advance in the efficiency of characterising first intron products. However, separate PCR and second intron product analysis, using agarose gel electrophoresis, was still required to resolve several common P. avium S-alleles.
Yamane et al. (2003b) demonstrated a method for the molecular typing of P. mume SFB genes to determine S-haplotype using genomic DNA blots and a PmSFB probe. Such a method could be employed in other Prunus species to determine S-haplotype. However, for large-scale population studies or analysis of extensive cultivar collections this would be impractical. A recent population study of wild P. lannesiana utilised genomic Southern analysis to assess three different RFLP patterns to determine S-genotype (Kato and Mukai 2004). This approach will have necessitated 435 restriction digests and subsequent Southern hybridisation to establish the S-genotype of the 145 individuals studied. The PCR method outlined here represents a significant advance and is eminently suited to undertaking large-scale population and cultivar surveys in cherry and other Prunus species. Not only can S-RNase and SFB alleles be quickly determined to classify the S-genotype but additionally we found that certain combinations of primers which amplify polymorphic P. avium simple sequence repeat (SSR) markers (Cipriani et al. 1999; Downey and Iezzoni 2000; Vaughan and Russell 2004) could be successfully combined into the same reaction. This allows individual accessions to be characterised on the basis of SSRs at the same time as S-genotype is determined.
The major limiting factor inhibiting research into the dynamics of self-incompatibility in natural populations of Prunus has been the lack of a fast and reliable method to determine S-haplotype in large numbers of individuals. We have subsequently employed the method described here to characterise a large wild population of P. avium with more than 1,500 individuals. This would have been expensive, time consuming and impractical using traditional methodologies. All of the SFB alleles reported here were reliably identified and correlated with their respective S-RNase allele intron products multiple times for numerous S-haplotypes. Furthermore, anomalous SFB 5′UTR intron/S-RNase intron profiles observed in some individuals drew our attention to several previously unreported S-haplotypes which were subsequently proven to contain new S-RNase alleles (Vaughan et al., in preparation).
The utility of SFB intron polymorphism demonstrated in P. avium SFB may well be applicable to other species. Qiao et al. (2004a) demonstrated that pollen behaviour in Petunia (Solanaceae) could be altered by the expression of an Antirrhinum (Scrophulariaceae) S-locus F-box protein. Thus it appears that SFB gene structure and function have remained conserved within these families since the GSI mechanism arose in the ancestral eudicots. The possible presence and utility of introns present in the 5′UTR of the F-box genes characterised within the Scrophulariaceae and Solanaceae has thus far not been investigated. However, within the Rosaceae, putative introns have been reported in the 5′UTR of P. dulcis SFBc (Ushijima et al. 2003) and recently for three P. armeniaca SFB alleles (Romero et al. 2004). Sequence analysis revealed that the conserved motifs observed in the P. avium SFB introns reported here are also present in these other species (data not shown). Furthermore, the putative P. armeniaca introns also exhibit sequence length polymorphism, 110, 106 and 137 bp for P. armeniaca SFB 1 , SFB 2 and SFB 4 introns, respectively. Preliminary research in our laboratories has shown that the primers presented here may be successfully employed in a number of other Prunus species including P. cerasifera, P. dulcis and P. tenella (data not shown) indicating that the 5′ intron border sequence and SFB gene sequence are conserved across these species and confirming the wider application of the methodology presented here to population studies in other species.
Abbreviations
- GSI:
-
Gametophytic self-incompatibility
- SI:
-
Self-incompatibilty
- SFB :
-
S-locus F-box
- SLFL :
-
Non-haplotype-specific S-locus F-box
References
Anderson MA, Cornish EC, Mau SL, Williams EG, Hoggart R, Atkinson A, Bonig I, Grego B, Simpson R, Roche PJ, Haley JD, Penschow JD, Niall HD, Tregear GW, Coghlan JP, Crawford RJ, Clarke AE (1986) Cloning of cDNA for a stylar glycoprotein associated with expression of self-incompatibility in Nicotiana alata. Nature 321:38–44
Bošković R, Tobutt KR (1996) Correlation of stylar ribonuclease zymograms with incompatibility alleles in sweet cherry. Euphytica 90:245–250
Bošković R, Tobutt KR (2001) Genotyping cherry cultivars assigned to incompatibility groups, by analysing stylar ribonucleases. Theor Appl Genet 103:475–485
Cipriani G, Lot G, Huang WG, Marrazzo MT, Peterlunger E, Testolin R (1999) AC/GT and AG/CT microsatellite repeats in peach Prunus persica (L) Batsch: isolation, characterisation and cross-species amplification in Prunus. Theor Appl Genet 99:65–72
Crane MB, Brown AG (1937) Incompatibility and sterility in the sweet cherry, Prunus avium L. J Pomol Hortic Sci 15:86–116
Crane MB, Lawrence WJC (1929) Genetical and cytological aspects of incompatibility and sterility in cultivated fruits. J Pomol Hortic Sci 7:276–301
De Cuyper B, Sonneveld T, Tobutt KR (2005) Determining self-incompatibility genotypes in Belgian wild cherries. Mol Ecol 14:945–955
Downey SL, Iezzoni AF (2000) Polymorphic DNA markers in black cherry (Prunus serotina) are identified using sequences from sweet cherry, peach and sour cherry. J Am Soc Hortic Sci 125:76–80
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Ducci F, Santi F (1998) The distribution of clones in managed and unmanaged populations of wild cherry (Prunus avium). Can J For Sci 27:1998–2004
Entani T, Iwano M, Shiba H, Che FS, Isogai A, Takayama S (2003) Comparative analysis of the self-incompatibility (S−) locus region of Prunus mume: identification of a pollen-expressed F-box gene with allelic diversity. Genes Cells 8:203–213
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Franklin-Tong N, Franklin FCH (2003) Gametophytic self-incompatibility inhibits pollen tube growth using different mechanisms. Trends Plant Sci 8:598–605
Igic B, Kohn JR (2001) Evolutionary relationships among self-incompatibility RNases. Proc Natl Acad Sci USA 98:13167–13171
Ikeda K, Igic B, Ushijima K, Yamane H, Hauck NR, Nakano R, Sassa H, Iezzoni AF, Kohn JR, Tao R (2004) Primary structural features of the S haplotype-specific F-box protein, SFB, in Prunus. Sex Plant Reprod 16:235–243
Ishimizu T, Endo T, Yamaguchi-Kabata Y, Nakamura KT, Sakiyama F, Norioka S (1998) Identification of regions in which positive selection may operate in S-RNase of Rosaceae, implication for S-allele-specific recognition sites in S-RNase. FEBS Lett 440:337–342
Kato S, Mukai Y (2004) Allelic diversity of S-RNase at the self-incompatibility locus in natural flowering cherry populations (Prunus lannesiana var. speciosa). Heredity 92:249–256
Lai Z, Ma WS, Han B, Liang LZ, Zhang YS, Hong GF, Xue YB (2002) An F-box gene linked to the self-incompatibility (S) locus of Antirrhinum is expressed specifically in pollen and tapetum. Plant Mol Biol 50:29–42
Matthews P (1970) Genetics and exploitation of self-fertility in the sweet cherry. In: Proceedings of the Angers Fruit Breed Symposium, INRA,Versailles, pp 307–316
McClure BA, Haring V, Ebert PR, Anderson MA, Simpson RJ, Sakiyama F, Clarke AE (1989) Style self-incompatibility gene-products of Nicotiana alata are ribonucleases. Nature 342:955–957
McCubbin AG, Kao TH (2000) Molecular recognition and response in pollen and pistil interactions. Annu Rev Cell Dev Biol 16:333–364
de Nettancourt D (2001) Incompatibility and incongruity in wild and cultivated plants. Springer, Berlin Heidelberg New York
Qiao H, Wang F, Zhao L, Zhou JL, Lai Z, Zhang YS, Robbins TP, Xue YB (2004a) The F-Box protein AhSLF-S-2 controls the pollen function of S-RNase-based self-incompatibility. Plant Cell 16:2307–2322
Qiao H, Wang HY, Zhao L, Zhou JL, Huang J, Zhang YS, Xue YB (2004b) The F-box protein AhSLF-S-2 physically interacts with S-RNases that may be inhibited by the ubiquitin/26S proteasome pathway of protein degradation during compatible pollination in Antirrhinum. Plant Cell 16:582–595
Romero C, Vilanova S, Burgos L, Martinez-Calvo J, Vicente M, Llacer G, Badenes ML (2004) Analysis of the S-locus structure in Prunus armeniaca L. Identification of S-haplotype specific S-RNase and F-box genes. Plant Mol Biol 56:145–157
Sassa H, Nishio T, Kowyama Y, Hirano H, Koba T, Ikehashi H (1996) Self incompatibility (S) alleles of the Rosaceae encode members of a distinct class of the T-2/S ribonuclease superfamily. Mol Gen Genet 250:547–557
Sijacic P, Wang X, Skirpan AL, Wang Y, Dowd PE, McCubbin AG, Huang S, Kao TH (2004) Identification of the pollen determinant of S-RNase-mediated self-incompatibility. Nature 429:302–305
Sonneveld T, Robbins TP, Boskovic R, Tobutt KR (2001) Cloning of six cherry self-incompatibility alleles and development of allele-specific PCR detection. Theor Appl Genet 102:1046–1055
Sonneveld T, Tobutt KR, Robbins TP (2003) Allele-specific PCR detection of sweet cherry self-incompatibility (S) alleles S1 to S16 using consensus and allele-specific primers. Theor Appl Genet 107:1059–1070
Sonneveld T, Tobutt KR, Vaughan SP, Robbins TP (2005a) Loss of pollen-S function in two self-compatible selections of Prunus avium is associated with deletion/mutation of an S-haplotype-specific F-box gene. Plant Cell 17:37–51
Sonneveld T, Robbins TP, Tobutt KR (2005b) Improved discrimination of self-incompatibility S-RNase alleles in cherry and high throughput genotyping by automated sizing of first intron PCR products. Plant Breed (in press)
Steinbachs JE, Holsinger KE (2002) S-RNase-mediated gametophytic self-incompatibility is ancestral in eudicots. Mol Biol Evol 19:825–829
Swofford DL (2003) PAUP* phylogenetic analysis using parsimony (* and other methods). Sinauer Associates, Sunderland
Tao R, Yamane H, Sugiura A, Murayama H, Sassa H, Mori H (1999) Molecular typing of S-alleles through identification, characterization and cDNA cloning for S-RNases in sweet cherry. J Am Soc Hortic Sci 124:224–233
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tobutt KR, Sonneveld T, Bekefi Z,Bošković R (2004) Cherry (in)compatibility genotypes—an updated cultivar table. Acta Hort 663:667–671
Ushijima K, Sassa H, Dandekar AM, Gradziel TM, Tao R, Hirano H (2003) Structural and transcriptional analysis of the self-incompatibility locus of almond: identification of a pollen-expressed F-box gene with haplotype-specific polymorphism. Plant Cell 15:771–781
Ushijima K, Yamane H, Watari A, Kakehi E, Ikeda K, Hauck NR, Iezzoni AF, Tao RT (2004) The S haplotype-specific F-box protein gene, SFB, is defective in self-compatible haplotypes of Prunus avium and P. mume. Plant J 39:573–586
Vaughan SP, Russell K (2004) Characterization of novel microsatellites and development of multiplex PCR for large-scale population studies in wild cherry, Prunus avium. Mol Ecol Notes 4:429–431
Wiersma PA, Wu Z, Zhou L, Hampson C, Kappel F (2001) Identification of new self-incompatibility alleles in sweet cherry (Prunus avium L.) and clarification of incompatibility groups by PCR and sequencing analysis. Theor Appl Genet 102:700–708
Xue YB, Carpenter R, Dickinson HG, Coen ES (1996) Origin of allelic diversity in Antirrhinum S locus RNases. Plant Cell 8:805–814
Yamane H, Ikeda K, Ushijima K, Sassa H, Tao R (2003a) A pollen-expressed gene for a novel protein with an F-box motif that is very tightly linked to a gene for S-RNase in two species of cherry, Prunus cerasus and P. avium. Plant Cell Physiol 44:764–769
Yamane H, Ushijima K, Sassa H, Tao R (2003b) The use of the S haplotype-specific F-box protein gene, SFB as a molecular marker for S-haplotypes and self-compatibility in Japanese apricot (Prunus mume). Theor Appl Genet 107:1357–1361
Acknowledgements
This research was supported by funding from Defra and the Forestry Commission (Grant no.WD0502). The authors would also like to thank Bart De Cuyper, Dr. Tineke Sonneveld and Dr. Radovan Bošković for provision of primers and P. avium material and for many helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Nybom
Rights and permissions
About this article
Cite this article
Vaughan, S.P., Russell, K., Sargent, D.J. et al. Isolation of S-locus F-box alleles in Prunus avium and their application in a novel method to determine self-incompatibility genotype. Theor Appl Genet 112, 856–866 (2006). https://doi.org/10.1007/s00122-005-0187-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-005-0187-9