
Abstract
The outbreak of novel coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) virus continually led to infect a large population worldwide. SARS-CoV-2 utilizes its NSP6 and Orf9c proteins to interact with sigma receptors that are implicated in lipid remodeling and ER stress response, to infect cells. The drugs targeting the sigma receptors, sigma-1 and sigma-2, have emerged as effective candidates to reduce viral infectivity, and some of them are in clinical trials against COVID-19. The antipsychotic drug, haloperidol, exerts remarkable antiviral activity, but, at the same time, the sigma-1 benzomorphan agonist, dextromethorphan, showed pro-viral activity. To explore the potential mechanisms of biased binding and activity of the two drugs, haloperidol and dextromethorphan towards NSP6, we herein utilized molecular docking–based molecular dynamics simulation studies. Our extensive analysis of the protein-drug interactions, structural and conformational dynamics, residual frustrations, and molecular switches of NSP6-drug complexes indicates that dextromethorphan binding leads to structural destabilization and increase in conformational dynamics and energetic frustrations. On the other hand, the strong binding of haloperidol leads to minimal structural and dynamical perturbations to NSP6. Thus, the structural insights of stronger binding affinity and favorable molecular interactions of haloperidol towards viral NSP6 suggests that haloperidol can be potentially explored as a candidate drug against COVID-19.
Key messages
•Inhibitors of sigma receptors are considered as potent drugs against COVID-19.
•Antipsychotic drug, haloperidol, binds strongly to NSP6 and induces the minimal changes in structure and dynamics of NSP6.
•Dextromethorphan, agonist of sigma receptors, binding leads to overall destabilization of NSP6.
•These two drugs bind with NSP6 differently and also induce differences in the structural and conformational changes that explain their different mechanisms of action.
•Haloperidol can be explored as a candidate drug against COVID-19.
Similar content being viewed by others

Introduction
The current outbreak of corona virus disease 2019 (COVID-19) caused by a novel coronavirus SARS-CoV-2 was first reported from Wuhan, China, in late December 2019 [1], which has subsequently affected the entire world, reporting nearly 26 million of confirmed cases of COVID-19 along with ~ 9.0-lakh deaths as per data recorded in September 1st week, 2020, posing a global threat for human health and economy. With so many novel studies and findings surfaced, since its inception, we are still lagging behind in development of an effective treatment strategy to control the virus spread and prevent the disease [2,3,4,5,6,7].
SARS-CoV-2 is an enveloped non-segmented large positive sense, single-stranded RNA virus (~ 30 kb) with 5′-cap structure and 3′-poly-A tail belonging to β-CoV category [8, 9]. Its RNA genome contains 29,891 nucleotides and encoding for ~ 9860 amino acids [9]. The genome codes for both structural proteins like spike (S), envelope (E), membrane (M), and nucleocapsid (N), along with many non-structural proteins (NSPs 1–16) [10]. While these NSPs linked to RNA replication and processing of subgenomic RNAs, the functions of some of the NSPs are not known. A key component, NSP6, is a membrane protein of approximately 34 kDa with eight transmembrane helices and a highly conserved C-terminus. Together with NSP3 and NSP4, NSP6 is involved in the formation of replication-transcription complexes (RTCs) or replication organelles (RO) by stimulating the rearrangement of host cell membranes [11]. These replication complexes serve many important functions during the virus life cycle and play an important role in infection [12, 13]. The expression of these three NSPs has been associated with the formation of different membranous structure characteristic of CoV-infected cells including the double-membrane vesicles (DMVs), large virion-containing vacuoles (LVCVs), cubic membrane structures (CMSs), and zippered endoplasmic reticulum (ER) spherules [14,15,16]. NSP6 protein is also involved in blocking ER-induced autophagosome/autolysosome vesicle formation that plays a protective role in checking viral production inside host cells. NSP6 through the activation of the omegasome pathway induces autophagy [17]. The autophagosomes produced by NSP6 are higher in number but smaller in size as compared with those induced by starvation [18]. This may favor coronavirus infection by limiting the ability of autophagosomes to deliver viral components to lysosomal degradation.
Recently, Gordon et al. [19] presented a blueprint of SARS-CoV-2-Human interactome to reveal potential drug targets against COVID-19. The authors identified 332 interactions between viral and host proteins, largely targeting the innate immune signaling pathway. Using this blueprint, they identified a series of drugs and compounds with high potential to fight COVID-19—some of which are now being entered into clinical trials. The authors found that SARS-CoV-2 NSP6 protein interacts with the sigma receptor, which regulates ER stress response [19] and blocks ER-induced autophagosome/autolysosome vesicle that restricts viral production. They found that drugs or molecules targeting sigma-1 and sigma-2 receptors had effectively inhibited virus replication and growth. These identified drugs or molecules include antipsychotics, haloperidol, and melperone, which are used to treat schizophrenia; antihistamines like clemastine and cloperastine; compound PB28; and the female hormone progesterone. Recently, Serkan Tulgar and co-authors [20] advocated the use of haloperidol, an anti-inflammatory agent, in preventing the progression and reduction of severity of COVID-19.
Gordon et al. [19] in the same study also identified the sigma-1 benzomorphan agonist, dextromethorphan, that actually has pro-viral activity, which helped the growth of the virus in cells. The authors suggested that dextromethorphan might activate sigma-1, which could help the activation of the stress response that the SARS-CoV-2 hijacks. Dextromethorphan is a common cough suppressant found in several over-the-counter cough medicines. It binds to NMDA receptors and can modulate glutamatergic signaling [21]. It also binds to serotonin transporters [22] and several other protein targets, including sigma-1 receptors [21, 22] which have been considered therapeutic targets for antidepressant drugs [23,24,25,26]. However, the Consumer Healthcare Products Association (CHPA) in their statement says that “There is currently no clinical data indicating that the cough suppressant dextromethorphan has a pro-viral effect in people with COVID-19 infection. The study results published by Gordon et al. [19] are experimental, preliminary, and not conclusive.”
More importantly, Theresa Enkirch et al. [27] had evaluated the anti-influenza activity of dextromethorphan in vitro, and in mice as well as in animal models. The authors demonstrated that dextromethorphan was able to inhibit viral replication both in vitro and in vivo. All these findings suggest that haloperidol and dextromethorphan are potential candidate binding to SARS-CoV-2 NSP6 and exert different viral activity. Haloperidol inhibits the SARS-CoV-2 with Ki of 2–12 nM, while dextromethorphan activates the virus with an activity constant, Ki of 118 nM [19]. However, it is still not clear about the theoretical basis of this contradictory and biased activity. To explore the molecular mechanism of antiviral activity of haloperidol and the pro-viral activity of dextromethorphan, a number of computational methods like molecular docking, all-atom molecular dynamics (MD) simulation, and the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM-PBSA) were employed in this study. These mechanism-relevant studies will explore the binding modes, examine the key residues in the binding process, and elucidate the detailed interaction mechanisms. The results are expected to provide insights into the binding mechanism of the NSP6-drug complexes, which will be useful for future exploration of efficient drug targets in COVID-19.
Methods
Structure modeling of SARS-CoV-2 NSP6
The three-dimensional structure of NSP6 was determined by the DeepMind algorithm AplhaFold, based on deep neural network learning [28]. The NSP6 model structure was energy-minimized to remove steric clashes, and the lowest energy structure was selected based on the MolProbity scores. In addition, the Zhang group also predicted models for the SARS-CoV-2 proteins [29] using the novel C-I-TASSER platform [30]. However, these models have poor local geometries, numerous atomic clashes, poor side-chain conformations, and bad backbone dihedral angles, as measured by the MolProbity score. The stereochemical quality of the modeled protein was evaluated using the structural analysis and verification server version 5.0 (SAVES) (https://servicesn.mbi.ucla.edu/SAVES/). This meta server runs six programs for checking the stereochemical quality of a protein structure by analyzing residue-by-residue geometry and overall structural geometry.
Molecular docking
The chemical structures of the drugs were downloaded from the ZINC Database [31] as MOL2 files and then converted into three-dimensional PDB files using Open Babel 2.4.1 suite. The Swiss-PDB tool was used to energy-minimize the protein structure to get the stable and low-energy conformation state of protein. All the docking studies have been performed with AutoDock Vina [32]. The AutoDock tools package [33] was used to prepare the protein structures for docking by adding hydrogen to the polar groups along with Kollman charges. Both the protein and ligand files were converted into PDBQT format using the AutoDock Vina plugin with scripts from the AutoDock Tools package. The AutoDock Vina generates up to 9 poses for each drug containing the molecular coordinates as well the Gibbs free energy variation (ΔG, kcal/mol) for each pose. Finally, the obtained top-posed docking conformations were subjected to post-docking energy minimization in Discovery Studio (DS 3.531). After docking, the resultant receptor-ligand complexes were visualized and studied by the Discovery Studio Visualizer (BIOVIA) and PyMOL [34], UCSF chimera 1.9 [35], and Ligplot+ [36].
Molecular dynamics simulation
The all-atom MD simulations were performed using Gromacs v5.1.4 on the atomic coordinates of SARS-CoV-2 NSP6, and NSP6 complexed with dextromethorphan and haloperidol. The force field was selected as CHARMM27, and the water model TIP3P was used to solvate the systems [37], and the ligand parameters were taken as described elsewhere [38]. The system was placed in the center of the octahedral simulation box with buffer distance (10 Å) and the water molecule padding around the system. To neutralize the system, 0.15-M counter ions (Na+ and Cl−) were added [39]. All the MD simulations were performed at physiological temperature, 300°K. The energy minimization of all three systems was performed using steepest descent as first, then conjugant gradients (50,000 steps for each). Bonds involving hydrogens were treated with SHAKE algorithm, PME (Particle mesh Ewald) was used to define long-range electrostatic forces, and PBC (periodic boundary condition) was applied to x, y, and z directions [40]. The ensemble processes, NVT and NPT, were applied for the equilibration of the system for the period of 500 ps. During the simulation, Berendsen thermostat [41] and Parrinello-Rahman pressure [42] were used to maintain the pressure and temperature, respectively. LINC algorithm was used to constrain the bonds and angles [43]. The van der Waals interactions were taken cared of by LJ potential with a cutoff of 0.10 nm. Using the NPT ensemble, production runs were performed for the period of 100 ns, with time integration. The energy, velocity, and trajectory were updated at the time interval of 10 ps. All production runs were done on CUDA-enabled Tesla GPU machine (DELL T640 with V100 GPU) and OS Centos 7 [44, 45]. Gromacs utilities and python scripts with MDTraj [46] were used to analyze the global structural order parameters, RMSD (root mean square deviation), Rg (radius of gyration), SASA (solvent accessible surface area), RMSF (root mean square fluctuation), PCA (principal component analysis), and free energy landscape (FEL), and DSSP was used to examine the secondary structure conformational dynamics during the simulation [47].
Ligand binding free energy estimation
We calculated the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM-PBSA) from the obtained MD trajectories of protein-ligand complexes to describe the structural stability of protein-ligand, spatial orientation ligands, and molecular interactions at binding site of NSP6. The MM-PBSA provides a robust estimation of free-binding energy, contacts, and the effect of solvent underlying the binding affinity of ligand molecules [48, 49]. The binding free energy is expressed as:
where Gcomplex represented the total free energy of protein-ligand complex, Gprotein as the free energy of protein, and Gligand used as the free energy of ligand. Neglecting entropy terms, the free energy for each entity can be represented as:
where ΔEMM is the average molecular mechanics interaction energy change (gas-phase) upon ligand binding. ΔGsolv is the solvation free energy change on ligand binding. EMM included the bonded, electrostatic, and van der Waals energy, and Gsolv included polar and non-polar solvation terms.
Residual local frustration analysis
We have applied the Fereiro and Woylness [50] algorithm for the calculation of residual frustration in the NSP6 structures. The residual frustration index was calculated from the Frustratometer server (http://www.frustratometer.tk) [51]. The results were explained based on the “single residual frustration index.” A frustration value (denoted as Z-score) greater than + 0.78 will be defined as minimally frustrated or stabilizing residue, while a frustration value less than − 0.78 will be defined as highly frustrated or destabilizing [52]. If the Z-score lies in between these two limits, the residue will be defined as neutral.
Results
Structure modeling of SARS-CoV-2 NSP6
The secondary structure of NSP6 was predicted using AlphaFold, a deep learning algorithm, and shown in Fig. 1. The constructed model of SARS-CoV-2 NSP6 has been verified through the SAVES v5.0 server. The Ramachandran plot showed that 91.7% and 8.3% of residues are located in the most favored, and additionally allowed, regions, respectively. The environmental profile for NSP6 was computed with Verify3D, which showed that almost 80% of the amino acids have scored more than zero. The non-bonded interactions between the atoms were also predicted with the ERRAT server, which showed the overall quality factor of 92.19 (Supplementary Figure S1). The overall three-dimensional structure of NSP6 (Fig. 1) consists of fourteen α-helices, a C-terminal, two antiparallel β-strands, and sixteen turns. The structure of NSP6 has eight transmembrane helices (TM1-TM8).

Three-dimensional model structure of SARS-CoV-2 NSP6 generated by AlphaFold. The transmembrane helices predicted through THHMM server
Binding site exploration through molecular docking analysis
To predict the binding site for performing the molecular docking, a ligand-independent binding site search was done on NSP6 using CASTp Server [53]. This program provides comprehensive and detailed quantitative characterization of geometric and topological features of protein structures. The server identifies a putative binding site with a solvent accessible surface area of 1076 Å2 and volume of 1442 Å3. This binding site of NSP6 is encompassed by helices H3 (residues 61-69), turn and coil region (residues 126-133) joining H6 and H7, H8-H9 (residues 170-179), and C-terminal H11-H12-β1 (residues 221-244) (Supplementary Figure S2). We have used the other two web-servers, PrankWeb [54] and COACH [55], for predicting the binding sites. These two servers also predict similar residues in the binding pocket, with certain changes.
For the molecular docking study, we utilized the AutoDock Vina, which is an open-source program and the most commonly used docking software with an effective scoring function [32]. Haloperidol is an antipsychotic drug and is an inhibitor of sigma-1, dopamine D2, and histamine H1 receptors; it inhibits SARS-CoV-2 with a Ki of 2–12 nM [19]. Dextromethorphan, an antidepressant and sigma-1 agonist, activates the virus with an activity constant, Ki of 118 nM [19]. The molecular docking results showed that both haloperidol and dextromethorphan form a good complex with NSP6 with docking (affinity) scores of − 7.7 kcal/mol and − 6.5 kcal/mol, respectively, which indicates KD (KD = expΔG/RT) values in the nanomolar to low micromolar range.
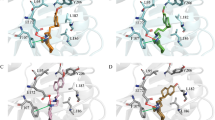
The binding mode for dextromethorphan based on the docking studies is presented in Fig. 2a. Dextromethorphan forms H-bonds with its methoxy group to Lys61 of NSP6. Both Cys229 and Tyr242 form a liophilic binding pocket with the formation of π-sulfur and π-alkyl interactions with aromatic rings. The morphian group binds with several hydrophobic residues, including His62, Arg233, Arg236, and Leu245, and forms additional van der Waals interactions from residues Asn232, Thr238, Asp243, and Pro282. This suggests that dextromethorphan-binding pocket lies mainly in C-terminal α-helix, H12, and β1 strand.

Molecular docking interactions of two drugs with SARS-CoV-2 NSP6 protein. Schematic representation of interactions made by a dextromethorphan and b haloperidol with NSP6 and their corresponding Ligplot+
The docking results show the different binding pose of haloperidol compared with dextromethorphan (Fig. 2b). Haloperidol is predicted to bind in the hydrophobic cavity formed by H7 (TM5), H9 (TM7), and C-terminal positively charged residues. The carbonyl oxygen of the phenone group forms an H-bond with Ser176, while the hydroxy group of piperidino forms H-bonds with Ser265. Due to the close proximity of Leu231, Leu237 and Leu239 show cation-π interactions with the chlorophenyl group of the drug and Ala136 form π-stacked interactions with the aromatic ketone of the phenone group. Additional van der Waals interactions from the residues Arg137, Trp140, Asn174, Tyr175 Val178, Thr238, and Gln290 are observed. Thus, a higher number of hydrogen bonds and hydrophobic interactions in haloperidol are observed in comparison to dextromethorphan, indicating the former as a good inhibitor molecule.
Evaluating the stability and conformational dynamics of NSP6-systems through MD simulation
To further characterize the binding-induced structural and conformational changes, all-atom MD simulations for 100 ns were performed to obtain the conformational sampling of the two systems. The selected docking conformations of NSP6 in complex with haloperidol and dextromethorphan were sampled by 100-ns MD simulation, and the dynamic stability of the complex was elucidated by calculating the Cα-RMSD values of the protein as the function of simulation time (Figure S3A). The RMSD trajectory indicated that all the systems are stable and well equilibrated after ~ 50 ns and well converged for further analysis. The RMSD plots showed that the NSP6 and NSP6-haloperidol remained mostly stable with a RMSD of ~ 0.5 nm, while NSP6-dextromethorphan showed a sharp increase in a RMSD at ~ 45 ns reaches up to 1.25 nm and then remained stable throughout the simulation. The result thus indicated that the NSP6-haloperidol complex showed almost similar RMSD as unliganded NSP6 and formed much more stable complex as compared with NSP6-dextromethorphan complex.
The analysis of the one-dimensional probability distribution function of the NSP6 without ligand showed a narrower distribution with a higher probability of 0.10 around the RMSD ≤ 5.0A and thus had a stable helical conformation (Fig. 3a). Indeed, in the presence of dextromethorphan, the complex showed a broader RMSD distribution with the appearance of new populations with the maximum probability of 0.12 at a RMSD of 1.25 nm, indicating a change in conformation leading to destabilization of α-helix of H12 (Fig. 3a). The Cα-RMSD beyond 0.12 nm indicates a transition from an α-helical conformation towards the extended configuration (Fig. 3a). However, the drug haloperidol showed a lower probability (P = 0.10) with a slightly broader distribution and a rightward shift of the RMSD population compared with the NSP6. At this RMSD, intrahelical H-bonds are still present to stabilize a helical conformation. Thus, dextromethorphan has highly unstable binding to NSP6 which is evident with the RMSD plot, while haloperidol binds strongly.

Probability distributions of structural parameters of NSP6 systems. a Cα-RMSD. b Radius of gyration (Rg). c SASA. d RMSF for NSP6 (green), NSP6-haloperidol (blue), and NSP6-dextromethorphan (red)
Rg is an effective parameter to evaluate the structural integrity and compactness of the studied systems. The Rg is defined as the mass-weighted root mean square distance of a collection of atoms from their common center of mass. The time evolution plot of Rg showed that all systems were compact, with the NSP6-dextromethorphan having the lowest Rg (Figure S3B). The Rg plot clearly suggests that the NSP6-dextromethorphan complex having the least Rg value forms a well compact complex than the NSP6-haloperidol complex. It also indicates that after binding of the drug, the NSP6 protein attains a compact conformation and the drug dextromethorphan could stay stably at the binding site, which provided a guarantee for stronger interaction between dextromethorphan and NSP6. The NSP6-haloperidol complex showed similar Rg to NSP6. The Rg differences between the NSP6 systems were minimal in haloperidol, indicating that haloperidol binding marginally changes the spatial packing of the residues. In case of the NSP6-dextromethorphan system, Rg distribution shows the mixture of two normal distributions that correctly describes the sampled configurations (Rg1 = 2.12 nm, P = 0.05 nm; Rg2 = 2.25 nm, P = 0.04 nm) (Fig. 3b). Interestingly, the first peak is slightly in lesser side than the NSP6 and NSP6-haloperidol systems, suggesting a more compact globular state. The second small peak falls near the NSP6-haloperidol system. A visual inspection revealed the more compact globular shape (Rg = ~ 1.65 nm) in the haloperidol system as a consequence of the lesser fluctuations (see RMSF results).
The SASA results showed stable trajectories without any fluctuations throughout the simulation, thus suggesting the structural stability of NSP6 in the presence of drugs (Figure S3C). The marginal increment of SASA in the NSP6-dextromethorphan system indicates increased exposure of the residues to the surface of NSP6 (Fig. 3c).
The RMSF displays the flexibility/mobility of each amino acid residue in the NSP6 and NSP6-drug complexes (Fig. 3d). It is worth noting that NSP6 alone showed lower RMSF (higher rigidity) in comparison with the complexes, maintaining lower flexibility in α-helices, except for the helix H5 (S89-D99). The NSP6-haloperidol complex does not increase the fluctuation and showed RMSF value similar to unliganded NSP6, suggesting a favorable protein-inhibitor association. From the figure, it can be observed that the RMSF values of residues 130-140 (correspond to H7) in NSP6-haloperidol were lower than unliganded NSP6, indicating the stabilization of these regions upon haloperidol binding. The residues 85-92 (correspond to H5), 96-110 (correspond to hinge between H5 and H6), and 245-260 (H13) showed a milder increase in fluctuations. However, dextromethorphan caused much higher fluctuations along the whole protein when bound to NSP6, indicating that dextromethorphan binding results in significant structural perturbation to NSP6 (Fig. 3d). The dextromethorphan binding, however, decreases the flexibility in residues 93, 94, 97, and 98 (correspond to H5), indicating the dominant role of these residues in drug binding.
From the RMSF analysis, we also observed that the NSP6-dextromethorphan complex showed maximum fluctuation in the C-terminal domain (residues 240-270 correspond to β1-H13-H14) while unliganded NSP6 and NSP6-haloperidol do not show this fluctuation. From the RMSF result, we thus conclude that the NSP6-haloperidol complex is more stable as compared with the NSP6-dextromethorphan complex. This has been further substantiated by the H-bonding analysis.
The protein-drug interaction is transient, and H-bonds play a critical role in the stability of the protein-drug complex. We have analyzed the number of H-bonds for the last 60-ns trajectories (Figure S4). The time evolution of H-bonds for the three systems has been monitored during the simulation and represented in Figure S4. The average number of H-bonds for the NSP6-haloperidol and NSP6-dextromethorphan complexes was 0-2 and 0-1, respectively. As shown in the figure, hydrogen bonding patterns in the NSP6-haloperidol system remained constant throughout the entire simulation. Whereas, hydrogen bond formation in the NSP6-dextromethorphan was unstable and absent most of the time during the simulation. For the last 20 ns of simulation, the number of intermolecular H-bonds is greater in the case of the NSP6-haloperidol system, indicating the most stable binding.
Secondary structure dynamics of NSP6-drug complexes
To further understand the drug binding-induced changes in secondary structure, the time evolution of the secondary structure profiles is shown in Fig. 4. As mentioned earlier, the NSP6 adopts a compact fold consisting of fourteen α-helices and two β-strands. Notable changes in the secondary structure profile were observed in the NSP6-drug complex. Most significant changes were an increase in helicity of the residues 89-99 (i.e., H5) and residues 200-205 (i.e., residues between H10 and H11). In the NSP6 and NSP6-drug complexes, the hinge region covering residues 100-110 can transform between helix or turn during simulation. In the case of the NSP6-haloperidol complex, the region covering residues 226-236 (i.e., H12) was lost to turn during the simulation. DSSP information indicates that dextromethorphan binding to NSP6 decreases the helicity of residues 150-160 (H7 and H8) while the H12 helix remains stable as in unliganded NSP6.

Secondary structures of NSP6 systems. Time evolution of the secondary structure profiles a NSP6, b NSP6-haloperidol, and c NSP6-dextromethorphan complexes
Evaluation of structural motions and conformational sampling in NSP6-drug complexes
Here, we use the principal component analysis (PCA) to examine the conformational sampling of the NSP6 systems via examining their dominant modes of motion. The covariance matrix of atomic fluctuations was diagonalized for predicting the eigenvalues. The first few eigenvectors play a key role in the motions of protein. It is apparent from the result that the first 5 eigenvectors have a larger eigenvalue for the unliganded than for the drug-bound NSP6, reflecting larger collective atomic fluctuations of the unliganded NSP6 (Figure S5). In addition, the first five principal components (PCs) account for more than 70% of motions observed for the last 40-ns trajectories of the NSP6 and NSP6-haloperidol systems, whereas only the first three PCs account for ~ 100% of the motions of the NSP6-dextromethorphan system (Figure S5). This indicates that the NSP6 and NSp6-haloperidol complex showed lesser motions as compared with the NSP6-dextromethorphan complex, and the first few PCs are not the same in the studied three systems.
The conformational sampling of the NSP6 systems in the essential subspace is shown in Figure 5A which illustrates the global motions along with the PC1 and PC2 projected by the Cα atom. The figure clearly indicates that modeled NSP6 showed a more stable cluster as compared with the NSP6-drug complex. At the same time, we found that the NSP6-dextromethorphan complex occupied a wide and different conformational subspace and showed fewer stable clusters as compared with NSP6-haloperidol. The PCA thus supports the findings that show the NSP6-dextromethorphan complex less stable with increased conformational dynamics.

Principal component analysis. a Projection of the motion of the protein in phase space along the PC1 and PC2 for over 100 ns of MD simulation at 300°K. b Average Eigen RMSF values for NSP6 systems were predicted for PC1. The color code for the figure is: NSP6 (black), NSP6-haloperidol (green), and NSP6-dextromethorphan (red)
To find the reasons how drug binding affects the motions described by PC1 and PC2, the displacements of PCs for the NSP6 systems were calculated and are shown in Fig. 5b. Average fluctuation for the NSP6-dextromethorphan in both the PCs were much higher as compared with NSP6-haloperidol and unliganded NSP6. This indicated that NSP6-dextromethorphan might influence the protein motions during binding, whereas the NSP6-haloperidol complex showed fewer motions during binding, which is consistent with the RMSF analysis (Fig. 3d).
To demonstrate the effects of drug binding on conformational redistributions, free energy landscapes (FEL) for the three systems were determined as a function of the top two principal components, PC1 and PC2. The 2D and 3D plots of the FEL are shown in Fig. 6. Each protein-drug complex has a different FEL pattern. FEL of NSP6 protein had multiple minima with small energy barriers in a single broad valley (basin). Most of the minima with the lowest energy had a flat end showing the clustering of low-energy conformations (Fig. 6a). After haloperidol binding, the basins segregate to the different coordinates and show clustered lowest energy minima close to each other. Some of the energy minima had a conical end suggesting the presence of stable conformation, while the minima with flat end indicating the absence of low-energy conformations (Fig. 6b). However, in the case of the dextromethorphan-bound NSP6, there exists only one deeper energy minima in the global free energy minimum region, indicating only one stable conformational state residing within this valley (basin) (Fig. 6c). The free energy valley is much deeper than the unliganded NSP6 and NSP6-haloperidol complex, indicating that the NSP6-dextromethorphan complex has a lower free energy value. Besides this, the NSP6-dextromethorphan complex also showed a reversed population shift relative to the unliganded NSP6.

The free energy landscape (FEL) of the simulated NSP6 systems based on the principal component analysis. a NSP6. b NSP6-haloperidol. c NSP6-dextromethorphan. The color bar represents the free energy value according to kcal mol−1. Dark blue spots indicate the energy minima and energetically favored protein conformations, and yellow spots indicate the unfavorable high-energy conformations
A comparison of the FELs for these two drug complexes of NSP6 reveals that the FEL of the haloperidol-bound NSP6 exhibits a more rugged free energy surface than that of the dextromethorphan-bound NSP6. Furthermore, the FEL of the NSP6-haloperidol complex contains a greater number of local free energy minima either in the global free energy minimum region (i.e., the funnel bottom) or in the region outside the global free minimum (i.e., the funnel wall), resulting in a more rugged and complex FEL.
Evaluation of interaction energetic features in NSP6-drug complexes
Evaluation of binding free energy is one of the important aspects of drug discovery as they describe the strength of non-bonded molecular associations during binding. The more negative free energy value signifies the stronger binding between the ligand and the protein. Using the MM-PBSA method [56, 57], binding free energy (ΔGbinding) was calculated as the difference between the free energy of the protein and protein-drug complex in the last 20 ns, sampled at 10-ps time intervals (Figure S6).
The average binding free energies and detailed energetic contribution components of the last 20 ns of MD trajectories were calculated and are shown in Table 1. It can be seen that haloperidol binds more strongly to NSP6 than dextromethorphan by − 12.74 kcal/mol, indicating enhanced binding affinity. While van der Waals energy (EvdW) largely contributed to the binding of haloperidol to NSP6, polar energy is unfavorable with the positive value. Besides, hydrophobic interaction played a crucial role in Hel binding to NSP6.
For dextromethorphan binding to NSP6, both polar energy and van der Waals interactions were favorable, while electrostatic energy is highly unfavorable with the positive value of 52.1 ± 3.39 kcal/mol (Table 1). According to Table 1, it is apparent that the polar (charged interactions) component was the key contributor in the binding free energy of the NSP6-dextromethorphan complex. The results indicated that the large unfavorable electrostatic term (ΔEele) was compensated by the large contribution of polar and van der Waals component in the binding process of dextromethorphan to NSP6. Therefore, the polar contribution was considered the main driving force in binding mechanism. To identify the key residues that contributed to the binding energy, we decomposed the binding energy at amino acid basis and the important ones with high contribution (≥ 0.1 kcal/mol) were identified and are shown in Fig. 7.

The per-residue binding free energy decomposition for the simulated NSP6-haloperidol (blue) and NSP6-dextromethorphan (red). The free energy values ≥ 0.1 kcal/mol contributes more to the binding interaction
As represented in Fig. 7, the values of amino acid energy with high contribution in each drug complex varied significantly. The decompositions of the relative binding energies of individual residues to the NSP6-haloperidol complex formation with the most favorable interactions were from Trp140, Trp165, Ala166, and Gly177, which comprised both charged and hydrophobic amino acids (Fig. 7). Residues within dextromethorphan-binding sites of NSP6 includes mainly charged amino acids like W31, E39, D112, D133, D134, D159, E195, D243, E250, and D267 (Fig. 7). Of note, the dextromethorphan interactions with NSP6, many charged residues like lysine (K61, 63, 109, 111, 151, 263, 270, 274, 281, and 285) and arginine (R93, 187, 233, 236, and 252), produced large positive unfavorable binding free energy values on average.
Thus, we have observed the large spatial shift of the binding residue clusters in both the drugs. Overall, haloperidol binds to the residue clusters near Trp165-Gly177 (while there was no energy contribution in the NSP6-dextromethorphan system). Also, the dextromethorphan binds to the C-terminal residue clusters Arg236-Asp267 (while there was no energy contribution in the NSP6-haloperidol system).
Dynamic cross-correlation matrix analysis
Calculation of correlation matrix is frequently utilized to illustrate dynamical information of proteins in two dimensions [52, 58, 59]. To observe the correlation in the dynamics, correlation matrices for each of the NSP6 systems were calculated through DynaMut web server [60] and are plotted in Fig. 8. The red regions in the correlation maps represent the strongly correlated motion of the residues while the blue regions are associated with strong anti-correlated motion of the residues. From the comparison of the correlation map of the NSP6 systems, substantial loss of motions in the correlation maps of the NSP6-drug complexes had been identified and labeled as shown in Fig. 8. In the presence of drugs, the helices of the hydrophobic core significantly destabilized and this is most apparent in the NSP6-dextromethorphan complex, whereby most of the helices showed the weakest correlation in motions compared with the NSP6-haloperidol complex.

Dynamic cross-correlation map (DCCM) of the Cα atoms around their mean positions computed through the DynaMut web server during the simulation. a NSP6. b NSP6-haloperidol. c NSP6-dextromethorphan complexes. The degrees of the correlation motions and anti-correlation motions are represented in blue and red, respectively. The degrees of motions correspond to the color bar
As can be seen, dextromethorphan binding leads to the complete loss of correlated motions of helices H9 and H11 in NSP6. The significant loss of correlated motions between H1-H6 and H1-H8 along with disruption of long-range motions between H5-H13, H6/H7-H11, and H5-β2, and short-range motions between H8/H9-H11 was observed. Interestingly, correlated motions between H9 and β2 have been observed in NSP6-dextromethorphan which is not present in the unliganded NSP6 protein. Moreover, the long-range anti-correlated motions between N- and C-terminal regions were also lost in the NSP6-dextromethorphan complex.
On the contrary, the NSP6-haloperidol complex displayed a somewhat better correlation in motion of residues of H8 and H11 compared with the NSP6-dextromethorphan system, hence indicating enhanced stability of the hydrophobic core (Fig. 8). However, similar to dextromethorphan, haloperidol binding also leads to complete loss of correlated motions between H1-H6, H1-H8, and H6/H7-H11, while retaining the motion between H8/H9-H11. There is also gain in long-range correlated motions between H2 and H13. Thus, the lack of correlation in the motion of the helical domains for the NSP6-dextromethorphan complex suggested the loss of contacts among these helices, which might cause the greater accessibility of the hydrophobic core to the solvent molecules, and subsequently the destabilization of the protein.
Residual frustration analysis
To examine the role of frustration in binding, we compute residual frustration in the protein by using the Protein Frustratometer server [51]. The frustration indices calculated through single residual frustration analysis indicate that SARS-CoV-2 NSP6 is more frustrated than a typical globular protein. Using the cutoff values of the frustration indices, we found that NSP6 protein is highly frustrated molecules with almost 27% of residues are highly frustrated (compared with 10% observed in a typical protein), and 36% of the residues are minimally frustrated (40% in general). The frustration values range between − 2.9 and + 1.4.
Here, the minimally frustrated residues were found in the helices H3, H5, H6, H10, and H11, whereas the highly frustrated residues were largely scattered and seen mainly in H5 and C-terminal H13 and H14 (Figure S7A–C).
Furthermore, we also monitored the frustration changes in NSP6 upon drug binding and the frustration profile showed some significant differences in protein-drug complex structure as shown in the Supporting Information Table S1.
As can be seen, dextromethorphan binding increased the local destabilization of the protein by increasing the highly frustrated residues (29.3%) while the local frustration is similar to apo NSP6 (35.8%). Haloperidol binding also increased the frustration to a little extent (28%) while keeping the minimal frustration unchanged (Table S1). The drug binding thus increases the number of highly frustrated residues and thus increases the local frustration and flexibility of the protein, especially in dextromethorphan binding.
Moreover, the residues involved in drug binding were dominantly neutral to minimal frustration. However, some of the dextromethorphan-binding residues like Ser32 and Leu276 gain frustration upon binding, whereas the frustration of Tyr132, Tyr136, Gly177, Arg236, and Lys270 decreased upon binding. Also, haloperidol binding increases the frustration of Leu231, Leu237, and Thr238 upon binding, and at the same time, it also decreased the frustration of residues like Tyr136, Phe184, and Phe269 (Table S1).
Moreover, to find out how drug binding induced the local frustration changes, we performed a comparative analysis of the spatial distribution of local frustrations mapped onto the secondary structure of the protein (Fig. 9). In particular, highly frustrated residues in the NSP6-dextromethorphan complex increased in the α-helix H1, H7, and H13, while the increase is seen in H11 for the NSP-haloperidol complex (Fig. 9a). The decrease in frustration was also seen for both the complexes mainly in C-terminal H11, H12, S1, and H14 for the NSP6-dextromethorphan complex, and for NSP6-haloperidol, a decrease is observed in H5, H8, and S1 (Fig. 9a). Additionally, the drug binding decreased the minimal frustration in H5, H7, H9, and H11 helices, and also increased the minimal frustration in H2 and H12 helices (Fig. 9b).

Frustration analysis in NSP6-drug complexes. The changes in residual frustration are distributed along the structural regions of NSP6. a The changes in minimal frustration values in NSP6-drug complex. b The changes in highly frustration values in NSP6-drug complex. The secondary structural regions are represented as: H-α-helix and S-β-strands
Thus, the coupling between structurally rigid C-terminal helices H10 and H11, and conformationally flexible helices, H5 and H13, is important for drug binding, and also the frustration index of the regions close to the binding site changes upon association.
Discussion
The all-atom MD simulation shows significant differences in the tertiary structure of the NSP6-drug complexes. The structural snapshots of the protein-drug complex were used to analyze the differences in tertiary structure caused by each drug (Figure S8).
Overall comparison of the binding conformations of haloperidol and dextromethorphan in NSP6
The NSP6-drug complexes show the significant differences in H2, H5, H7, and C-terminal regions comprising H12, H13, β1, and β2 (Figure S8). The haloperidol and dextromethorphan complexes have a kink in H2, H5, and H7 at midway of simulation (50 ns) that causes a change in orientation when the drug binds (Figure S8 A, C). The dextromethorphan binding induces much larger kink at H2 and H5 with a much larger deviation also (RMSD = 3.59) (Figure S8C). At the end of the simulation, the haloperidol and dextromethorphan complexes showed an increase in twist of H12 and H13, and both the strands of the C-termini along with the kinks present in H2, H5, and H7 (Figure S8B, D). Similar to what was observed at ~ 50 ns, dextromethorphan binding induces much larger disruption (RMSD = 3.24). Thus, although both drugs lead to bending of the helices, dextromethorphan binding induces larger disruption of the helices and thus more destabilization observed in the protein.
The protein-drug binding analysis shows that dextromethorphan and haloperidol interact with many key residues and are shared between both the drugs. The residues interacting with each drug during the simulation were compared and visual 2D representations are shown in Figures S9 and S10.
Figure 2 a shows the initial pose of the dextromethorphan molecule in the MD simulation, which is also the best pose from the docking study. As can be seen, the dextromethorphan molecule formed hydrogen bonds with residue Lys 61, and several van der Waals interactions with His62, Asn232, Arg233, Arg236, Thr238, Asp243, Leu245, and Pro282. Figure 2 b shows the haloperidol binding sites in NSP6 which is comprised of helix H7, and H9 residues (i.e., H7: Tyr132, Asp134, Ala136, Arg137, Trp140; H9: Asn174, Tyr175, Ser176, and Val178) and C-terminal positively charged residues Leu231, Leu237, Leu239, and Ser265.
The structural snapshots of the drug-protein complexes have been shown in Fig. 10. It has been seen during simulation that dextromethorphan has drifted to the different locations inside the NSP6 during ~ 20–100 ns of simulation. In the first ∼ 20 ns, dextromethorphan stayed in its initial location (Fig. 10a). After that, it drifted away from its initial location and moved into the water above the binding pocket and the interaction becomes minimal, only with Tyr234, Phe235, and Leu276 (Fig. 10b). Without drifting further into the water, the dextromethorphan molecule re-entered to the enlarged binding pocket, where it interacts mainly with H12 residues (Phe225, Leu230, Arg233, Tyr234, and Arg236) through the hydrophobic interaction (Fig. 10c).

The structural snapshots of NSP6-drug complex observed during the MD simulation (0 ns, 25 ns, 50 ns, 100 ns) for the most abundant structure of a–d NSP6-dextromethorphan and e–h NSP6-haloperidol
During the second half of the MD simulation (~ 50–100 ns), the dextromethorphan molecule stably remained at a new binding pose with much stronger interactions. At this new location, dextromethorphan coordinates with Ser32 through H-bonds, and hydrophobic interactions by Trp31, Ile189, Val190, Met192, Cys193, Phe200, and Phe201 (H9-H10) (Fig. 10d).
In contrast, the drug haloperidol was stably bound inside the NSP6 pocket and explored two binding poses during the simulation. For the first 50 ns, it remained at its initial docking pose where it forms H-bonds to Ser 176 and Ser265, and several hydrophobic interactions mediated by Arg137, Trp140, Asn174, Tyr175, Val178, and Thr238 (Fig. 10e, f). After that, it drifted to another binding site and remained stable there, until the end of the simulation. At this new site, haloperidol interacts mainly with H7-H9 residues (Trp140, Asn144, Trp165, Ala166, Ile169, Ser170, Ser176, Val178, and Phe184), Leu237, Leu239, and Ile266, Phe269 (H14) (Fig. 10g, h).
Conclusion
To better understand the mechanism of biased activity of SARS-CoV-2 in the presence of two sigma-R1 binding drugs to NSP6, molecular dynamics simulation studies were employed. Our data suggests dextromethorphan binds to C-terminal helices while haloperidol binding sites are in the middle of the protein domain in helices H7 and H9. The disruption of the alpha helix H7 and H8 is significant for the dextromethorphan complex along with a large kink in H5 and H7. The NSP6-haloperidol complex showed a less significant change in the tertiary structure despite major disruption of the H12 helix.
Furthermore, the analyses of RMSD, RMSF, PCA, FEL, and dynamic cross-correlation matrix (DCCM) indicated that dextromethorphan binding leads to destabilization of the protein with the loss of correlated motions and residual frustrations. In contrast, haloperidol binding brings milder alterations in these order parameters and thus showed minimal changes in stability compared with dextromethorphan system. Besides, intermolecular hydrogen bonds were constantly formed in haloperidol with high occupation indicating the more stabilization of the NSP6-haloperidol system. In conclusion, the study elucidated the detailed interaction mechanism of dextromethorphan and haloperidol to NSP6 protein and the associated structural and dynamical changes upon drug binding. These results will significantly enhance our understanding of the working mode of these drugs at the molecular and structural level and will contribute to the future rational drug design for COVID-19.
References
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y et al (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395:507–513
Hossein-Khannazer N, Shokoohian B, Shpichka A, Aghdaei HA, Timashev P, Vosough M (2020) Novel therapeutic approaches for treatment of COVID-19. J Mol Med (Berl) 98:789–803
Touma M (2020) COVID-19: molecular diagnostics overview. J Mol Med (Berl) 98:947–954
Olwenyi OA, Dyavar SR, Acharya A, Podany AT, Fletcher CV, Ng CL, Reid SP, Byrareddy SN (2020) Immuno-epidemiology and pathophysiology of coronavirus disease 2019 (COVID-19). J Mol Med (Berl). https://doi.org/10.1007/s00109-020-01961-4
Comentale G, Manzo R, Pilato E (2020) Sars-Cov-2 interference in HEME production: is it the time for an early predictive biomarker? J Mol Med (Berl) 98:1053–1054
Khatoon F, Prasad K, Kumar V (2020) Neurological manifestations of COVID-19: available evidences and a new paradigm. J Neurovirol. https://doi.org/10.1007/s13365-020-00895-4
Kalita P, Padhi AK, Zhang KYJ, Tripathi T (2020) Design of a peptide-based subunit vaccine against novel coronavirus SARS-CoV-2. Microb Pathog 145:104236
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R et al (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733
Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R (2020) Features, evaluation, and treatment of coronavirus (COVID-19). DOI NBK554776 [bookaccession]
Barnard DL (2008) Coronaviruses: molecular and cellular biology. Futur Virol 3:119–123
van der Hoeven B, Oudshoorn D, Koster AJ, Snijder EJ, Kikkert M, Barcena M (2016) Biogenesis and architecture of arterivirus replication organelles. Virus Res 220:70–90
Zalinger ZB, Elliott R, Rose KM, Weiss SR (2015) MDA5 is critical to host defense during infection with murine coronavirus. J Virol 89:12330–12340
He Y, Xie Z, Dai J, Cao Y, Hou J, Zheng Y, Wei T, Mo M, Wei P (2016) Responses of the Toll-like receptor and melanoma differentiation-associated protein 5 signaling pathways to avian infectious bronchitis virus infection in chicks. Virol Sin 31:57–68
Oudshoorn D, Rijs K, Limpens R, Groen K, Koster AJ, Snijder EJ, Kikkert M, Barcena M (2017). Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. mBio 8. DOI https://doi.org/10.1128/mBio.01658-17
Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ (2008) SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6:e226
Maier HJ, Hawes PC, Cottam EM, Mantell J, Verkade P, Monaghan P, Wileman T, Britton P (2013) Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. mBio 4: e00801-e00813. DOI
Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, Britton P, Ktistakis NT, Wileman T (2011) Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 7:1335–1347
Cottam EM, Whelband MC, Wileman T (2014) Coronavirus NSP6 restricts autophagosome expansion. Autophagy 10:1426–1441
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL et al (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. https://doi.org/10.1038/s41586-020-2286-9
Tulgar S, Ahiskalioglu A, Kok A, Thomas DT (2020) Possible old drugs for repositioning in COVID-19 treatment: combating cytokine storms from haloperidol to anti-interleukin agents. Turk J Anaesthesiol Reanim 48:256–257
Lauterbach EC (2012) An extension of hypotheses regarding rapid-acting, treatment-refractory, and conventional antidepressant activity of dextromethorphan and dextrorphan. Med Hypotheses 78:693–702
Werling LL, Keller A, Frank JG, Nuwayhid SJ (2007) A comparison of the binding profiles of dextromethorphan, memantine, fluoxetine and amitriptyline: treatment of involuntary emotional expression disorder. Exp Neurol 207:248–257
Matsumoto RR, Nguyen L, Kaushal N, Robson MJ (2014) Sigma (sigma) receptors as potential therapeutic targets to mitigate psychostimulant effects. Adv Pharmacol 69:323–386
Fishback JA, Robson MJ, Xu YT, Matsumoto RR (2010) Sigma receptors: potential targets for a new class of antidepressant drug. Pharmacol Ther 127:271–282
Nguyen L, Robson MJ, Healy JR, Scandinaro AL, Matsumoto RR (2014) Involvement of sigma-1 receptors in the antidepressant-like effects of dextromethorphan. PLoS One 9:e89985
Prasad K, Khatoon F, Rashid S, Ali N, AlAsmari AF, Ahmed MZ, Alqahtani AS, Alqahtani MS, Kumar V (2020) Targeting hub genes and pathways of innate immune response in COVID-19: a network biology perspective. Int J Biol Macromol. https://doi.org/10.1016/j.ijbiomac.2020.06.228
Enkirch T, Sauber S, Anderson DE, Gan ES, Kenanov D, Maurer-Stroh S, von Messling V (2019) Identification and in vivo efficacy assessment of approved orally bioavailable human host protein-targeting drugs with broad anti-influenza A activity. Front Immunol 10:1097
Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Zidek A, Nelson AWR, Bridgland A et al (2020) Improved protein structure prediction using potentials from deep learning. Nature 577:706–710
Zhang C, Zheng W, Huang X, Bell EW, Zhou X, Zhang Y (2020) Protein structure and sequence reanalysis of 2019-nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV-1. J Proteome Res 19:1351–1360
Zheng W, Li Y, Zhang C, Pearce R, Mortuza SM, Zhang Y (2019) Deep-learning contact-map guided protein structure prediction in CASP13. Proteins 87:1149–1164
Sterling T, Irwin JJ (2015) ZINC 15--ligand discovery for everyone. J Chem Inf Model 55:2324–2337
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Seeliger D, de Groot BL (2010) Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des 24:417–422
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51:2778–2786
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2:19–25
Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011) SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32:2359–2368
Joung IS, Cheatham TE 3rd (2008) Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B 112:9020–9041
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Berendsen HJC, Grigera JR, Straatsma TP (1987) The missing term in effective pair potentials. J Phys Chem 91:6269–6271
Parrinello M, Rahman A (1980) Crystal structure and pair potentials: a molecular-dynamics study. Phys Rev Lett 45:1196–1199
Berk H, Henk B, C. BHJ, M. FJGE (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18: 1463-1472. DOI:
Kumar V, Pandey P, Idrees D, Prakash A, Lynn AM (2019) Delineating the effect of mutations on the conformational dynamics of N-terminal domain of TDP-43. Biophys Chem 250:106174
Prakash A, Kumar V, Banerjee A, Lynn AM, Prasad R (2020) Structural heterogeneity in RNA recognition motif 2 (RRM2) of TAR DNA-binding protein 43 (TDP-43): clue to amyotrophic lateral sclerosis. J Biomol Struct Dyn:1–11. https://doi.org/10.1080/07391102.2020.1714481
McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernandez CX, Schwantes CR, Wang LP, Lane TJ, Pande VS (2015) MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J 109:1528–1532
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637
Kumar N, Srivastava R, Prakash A, Lynn AM (2019) Structure-based virtual screening, molecular dynamics simulation and MM-PBSA toward identifying the inhibitors for two-component regulatory system protein NarL of Mycobacterium Tuberculosis. J Biomol Struct Dyn:1–15. https://doi.org/10.1080/07391102.2019.1657499
Pandey P, Lynn AM, Bandyopadhyay P (2017) Identification of inhibitors against alpha-isopropylmalate synthase of mycobacterium tuberculosis using docking-MM/PBSA hybrid approach. Bioinformation 13:144–148
Ferreiro DU, Hegler JA, Komives EA, Wolynes PG (2007) Localizing frustration in native proteins and protein assemblies. Proc Natl Acad Sci U S A 104:19819–19824
Parra RG, Schafer NP, Radusky LG, Tsai MY, Guzovsky AB, Wolynes PG, Ferreiro DU (2016) Protein Frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res 44:W356–W360
Husain S, Kumar V, Hassan MI (2018) Phosphorylation-induced changes in the energetic frustration in human Tank binding kinase 1. J Theor Biol 449:14–22
Tian W, Chen C, Lei X, Zhao J, Liang J (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46:W363–W367
Jendele L, Krivak R, Skoda P, Novotny M, Hoksza D (2019) PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res 47:W345–W349
Yang J, Roy A, Zhang Y (2013) Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 29:2588–2595
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W et al (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33:889–897
Kumari R, Kumar R, Lynn A (2014) g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54:1951–1962
Chen J, Wang J, Zhu W, Li G (2013) A computational analysis of binding modes and conformation changes of MDM2 induced by p53 and inhibitor bindings. J Comput Aided Mol Des 27:965–974
Chen J, Liang Z, Wang W, Yi C, Zhang S, Zhang Q (2014) Revealing origin of decrease in potency of darunavir and amprenavir against HIV-2 relative to HIV-1 protease by molecular dynamics simulations. Sci Rep 4:6872
Rodrigues CH, Pires DE, Ascher DB (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 46:W350–W355
Acknowledgments
The authors sincerely thank the Amity University, Noida, for providing facilities. Authors gratefully acknowledge the computational facility funded by Science and Engineering Research Board (SERB), Government of India (Ref. No.: YSS/2015/000228/LS).
Author information
Authors and Affiliations
Contributions
V.K. designed the study. P.P. and K.P. performed the experiments and calculations. V.K. and A.P. analyzed the data. P.P. and K.P. prepared figures of the results. V.K. wrote the manuscript with the contributions of K.P. and A.P.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pandey, P., Prasad, K., Prakash, A. et al. Insights into the biased activity of dextromethorphan and haloperidol towards SARS-CoV-2 NSP6: in silico binding mechanistic analysis. J Mol Med 98, 1659–1673 (2020). https://doi.org/10.1007/s00109-020-01980-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-020-01980-1