
Abstract
Cardiovascular disease is among the main causes of morbidity and mortality in developed countries. The pathological process of the heart is associated with altered expression profile of genes that are important for cardiac function. MicroRNAs (miRNAs) have emerged as one of the central players of gene expression regulation. The implications of miRNAs in the pathological process of cardiovascular system have recently been recognized, representing the most rapidly evolving research field. Here, we summarize and analyze the currently available data from our own laboratory and other groups, providing a comprehensive overview of miRNA function in the heart, including a brief introduction of miRNA biology, expression profile of miRNAs in cardiac tissue, role of miRNAs in cardiac hypertrophy and heart failure, the arrhythmogenic potential of miRNAs, the involvement of miRNAs in vascular angiogenesis, and regulation of cardiomyocyte apoptosis by miRNAs. The target genes and signaling pathways linking the miRNAs to cardiovascular disease are highlighted. The applications of miRNA interference technologies for manipulating miRNA expression, stability, and function as new strategies for molecular therapy of human disease are evaluated. Finally, some specific issues related to future directions of the research on miRNAs relevant to cardiovascular disease are pinpointed and speculated.
Similar content being viewed by others
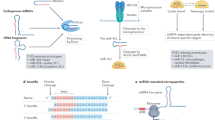


Introduction
MicroRNAs (miRNAs) have emerged as one of the central players of gene expression regulation. While the role of miRNAs in oncogenesis and cardiac development has been well appreciated over the past few years, the implications of miRNAs in the pathological process of cardiovascular system have only been recognized very recently [1–6]. The research on miRNAs in relation to cardiovascular disease has become a most rapidly evolving field. Several excellent review articles and editorial commentaries on this subject have been published lately [1–6]. Here, we do not attempt to repeat the contents and thoughts appearing in these papers. Instead, we try to summarize and analyze the currently available data from our own laboratory and other groups, providing a comprehensive overview of miRNAs and cardiovascular disease. The functional role of miRNAs in cardiac development and cardiovascular disease of adult hearts (including cardiac hypertrophy, heart failure, arrhythmias, cardiomyocyte apoptosis, vascular angiogenesis) and the target genes and signaling pathways linking these miRNAs to the events will be discussed. The applications of miRNA interference technologies for manipulating miRNA expression, stability, and function as new strategies for molecular therapy of human disease will be evaluated. Finally, some specific issues related to future directions of the research on miRNAs relevant to cardiovascular disease will be pinpointed and speculated [1–6].
miRNA biology
miRNAs are initially transcribed as long RNA precursors called primary miRNAs that requires the RNase III enzyme Drosha in the nucleus to be trimmed into premature miRNAs. The latter precursor, characterized by a 33-nucleotide (nt) stem-loop or hairpin structure of 60–70 nts, are exported to the cytoplasm where they are subsequently cropped to become mature miRNAs of 21–26 nts in length, by another RNase III enzyme Dicer [7–11].
Mature miRNAs can interact with Argonaute to form the RNA-induced silencing complex (RISC) and then guide the RISC to their target mRNAs, most favorably to the 3'-untranslated region (3'UTR). In order for a miRNA to produce functional consequences, its 5'-end 7 to 8 nts must have exact base pairing to the target mRNA, the “seed” region, and partial complementarity with the rest of the sequence. An miRNA can either inhibit translation or induce degradation of its target mRNA, primarily depending upon the overall degree of complementarity of the binding site, number of binding sites, and the accessibility of the bindings sites (determined by free energy states). The greater the complementarity of the accessible binding sites, the more likely an miRNA degrades its targeted mRNA, and those miRNAs that display imperfect sequence complementarities with target mRNAs primarily result in translational inhibition [12–17]. The more accessible the binding sites for an miRNA, the greater the actions of that miRNA owing to the potential cooperative miRNA–mRNA interactions from different sites. It is noteworthy that the finding by a most recent study that miRNAs can also act to enhance translation when AU-rich elements and miRNA target sites coexist at proximity in the target mRNA and when the cells are in the state of cell-cycle arrest [18]. It is proposed that translation regulation by miRNAs oscillates between repression and activation during the cell cycle.
MiRNAs are an abundant RNA species constituting >3% of the predicted human genes, which regulates ~30% of protein-coding genes [19]. The high sequence conservation across metazoan species suggests strong evolutionary pressure and participation of miRNAs in essential biologic processes such as cell proliferation, differentiation, apoptosis, metabolism, stress, etc. [20, 21]. It has been predicted that each single miRNA can have >1,000 target genes and each single protein-coding gene can be regulated by multiple miRNAs [22, 23]. This is simply because the actions of miRNAs are sequence or motif specific but not gene specific; different genes can have similar binding motifs for a given miRNA and a given gene can have the binding motifs for distinct miRNAs.
Expression profile of miRNAs in the heart
The expression profile of miRNAs appears to be tissue–cell specific. This implies that a group of miRNAs express only in a certain tissue–cell type or different groups of miRNAs expressed in different tissues–cells. For example, the miRNA expression profile in artery is different from that in heart. The most abundant miRNAs in cardiac muscles are miR-1, let-7, miR-133, miR-126-3p, miR-30c, and miR-26a [24]. However, in artery smooth muscles, the most abundant miRNAs are miR-145, let-7, miR-125b, miR-125a, miR-23, and miR-143, though miR-1 and miR-133 are also expressed in artery smooth muscles [25]. The differential tissue distributions of miRNAs suggest tissue- or even cell-type-specific function of these molecules.
The expression profile of miRNAs is also disease status dependent. A particular pathological process is associated with the expression of a particular group of miRNAs, a signature expression pattern of miRNAs. These signature patterns could aid in the diagnosis and prognosis of human disease. This notion has been supported by studies on cardiovascular disease, human cancer, and other disorders.
Among >500 miRNAs identified thus far in mammalians, miR-1, miR-133, and miR-208 are considered muscle specific, being primarily expressed in cardiac and skeletal muscles [17, 26–32]. The miR-1 family is comprised of the miR-1 subfamily and miR-206, with the former subfamily consisting of two transcripts, miR-1-1 and miR-1-2, that possess an identical mature sequence but are encoded by distinct genes located on chromosomes 2 and 18, respectively. The miR-133 family is comprised of miR-133a-1, miR-133a-2, and miR-133b, with miR-133a expressed from bicistronic units together with the miR-1 subfamily and miR-133b together with miR-206 [26, 27]. The resulting mature products from the miR-133 family are either identical or have only 1-nt difference. miR-208 is encoded by an intron in the α-myosin heavy chain (MHC) gene [32]. Cardiac-muscle-specific expressions of miR-1 and miR-133 are controlled by serum responsive factor (SRF)–myocardin and of miR-208 are associated with expression of α-MHC gene. A recent study indeed confirmed that >90% of miR-1 transcripts were blocked in the SRF knockout hearts [33]. By comparison, skeletal-muscle-specific expressions of miR-1 and miR-133 are controlled by myogenic differentiation–myocyte enhancer factor 2 (MEF2). A most recent study further revealed that MEF2 directly activates transcription of miR-1-2 and miR-133a-1 cluster and of miR-1-1 and miR-133a-2 locus as well [34].These data suggest that the tissue-specific expressions of miRNAs are largely determined by transcription factors.
miRNAs and cardiac development
Heart malformations occur in as high as 1% of newborns, presenting a significant clinical problem in our modern world. The muscle-specific miRNAs are expressed in the early stages during cardiogenesis. Microarray analysis revealed an increased expression of miR-1 and miR-133 in developing mouse hearts from embryonic day (E)12.5 through to at least E18.5, indicating a requirement of these miRNAs for cardiac development [26]. Several lines of evidence have been obtained in support of this notion [17, 26].
Block of miRNA biogenesis by tissue-specific deletion of Dicer causes lethality of embryos due to defects in cardiogenesis. When Dicer was efficiently deleted in the heart, death of the embryos by E12.5 due to cardiac failure was consistently observed [29]. Dicer-deficient embryos developed cardiac failure due to pericardial edema and underdevelopment of the ventricular myocardium. These phenotypes are in agreement with the defects of heart development in zebrafish embryos devoid of Dicer function [35].
Studies using both “gain-of-function” and “loss-of-function” approaches point to the roles of miR-1 and miR-133 in modulating cardiogenesis and in maintenance of muscle–gene expression. Genetic engineering in Drosophila demonstrated that miR-1 may “fine-tune” Notch ligand Delta that is critically involved in differentiation of cardiac and somatic muscle progenitors and targets a pathway required for progenitor cell specification and asymmetric cell division [28]. In Xenopus laevis, injection of either miR-1 at one-cell stage results in complete absence of cardiac tissue [26]. On the other hand, introduction of miR-133 allows cardiac tissue to form, but the tissue is disorganized and does not lead to chamber formation. The authors of this study proposed that both miR-1 and miR-133 are required for proper heart development and they may have distinct roles with miR-1 promoting myogenesis while miR-133 enhancing myoblast proliferation.
In mouse, miR-1 is responsible for the inhibition of cardiomyocyte progenitor proliferation via inhibition of translation of Hand2 [17], a transcription factor known to regulate ventricular cardiomyocyte expansion [36]. Overexpression of miR-1 in a transgenic mouse model resulted in a phenotype characterized by thin-walled ventricles, attributable to premature differentiation and early withdrawal of cardiomyocytes from the cell cycle [29]. In contrast, many of the embryos from miR-1-2 knockout mice demonstrated ventricular septal defects in a subset that suffer early lethality. The adult miR-1-2-deficient mice had thickened chamber walls attributable to hyperplasia of the heart [29].
miRNAs and cardiac hypertrophy and heart failure
In response to stress (such as hemodynamic alterations associated with myocardial infarction, hypertension, aortic stenosis, valvular dysfunction, etc), the adult heart undergoes remodeling process and hypertrophic growth to adapt to altered workloads and to compensate for the impaired cardiac function. Hypertrophic growth manifests enlargement of cardiomyocyte size and enhancement of protein synthesis through the activation of intracellular signaling pathways and transcriptional mediators in cardiac myocytes. The process is characterized by a reprogramming of cardiac gene expression and the activation of “fetal” cardiac genes [37]. In light of the role of miRNAs in cardiac development, one has a good reason to believe an involvement of miRNAs in cardiac hypertrophy. It is thus not surprising to see that this topic has indeed become an intensively studied subject; within a year from late 2006 up to now, there have been seven published original research articles focused on miRNAs and cardiac hypertrophy–heart failure. The findings from these studies reveal an important role for specific miRNAs in the control of hypertrophic growth and chamber remodeling of the heart and point to miRNAs as potential therapeutic targets in heart disease. These studies reported some common findings but also presented some controversies.
The first common finding is that an array of miRNAs is significantly altered in their expression, either upregulated or downregulated, and studies from different research groups demonstrated overlapping miRNAs that are altered in cardiac hypertrophy. The second common finding is that single miRNAs can critically determine the progression of cardiac hypertrophy. Olson's [38] group reported >12 miRNAs that are upregulated or downregulated in cardiac tissue from mice in response to transverse aortic constriction (TAC) or expression of activated calcineurin, stimuli that induce pathological cardiac remodeling. Many of these miRNAs were found similarly regulated in failing human hearts. Forced overexpression of stress-inducible miRNAs induced hypertrophy in cultured cardiomyocytes. Particularly, overexpression of miR-195 alone, which was upregulated during cardiac hypertrophy, was sufficient to induce pathological cardiac growth and heart failure in transgenic mice. However, the target genes for miR-195 relevant to hypertrophy have not been studied. The same group recently found that miR-208, encoded by an intron of the α-MHC gene, is required for cardiomyocyte hypertrophy, fibrosis, and expression of β-MHC in response to stress and hypothyroidism [32]. The study showed that miR-208 mutant mice failed to undergo stress-induced cardiac remodeling, hypertrophic growth, and β-MHC upregulation, whereas transgenic expression of miR-208 was sufficient to induce β-MHC. miR-208 regulates β-MHC by repressing the thyroid hormone receptor associated protein 1, a cofactor of the thyroid hormone receptor and a predicted miR-208 target mRNA. Abdellatif's [39] group reported an array of miRNAs that are differentially and temporally regulated during cardiac hypertrophy. They found that miR-1 was singularly downregulated as early as day 1, persisting through day 7, after TAC-induced hypertrophy in a mouse model. Overexpression of miR-1 carried by adenovirus vector inhibited its in silico-predicted growth-related targets, including Ras guanosine-triphosphatase-activating protein, cyclin-dependent kinase 9, fibronectin, and Ras homolog enriched in brain, in addition to protein synthesis and cell size. Their study also suggests that miRNA expression profiles at different time points after TAC are different, with expression of >50 miRNAs progressively changing during development of pressure overload cardiac hypertrophy. Thus, they proposed that miRNAs play an essential regulatory role in the development of cardiac hypertrophy, wherein downregulation of miR-1 is necessary for the relief of growth-related target genes from its repressive influence and induction of hypertrophy. A study from Condorelli's group focuses on the role of miR-133 and miR-1 in cardiac hypertrophy with three murine models: TAC mice, transgenic mice with selective cardiac overexpression of a constitutively active mutant of the Akt kinase, and human tissues from patients with cardiac hypertrophy [40]. They first showed that cardiac hypertrophy in all three models resulted in reduced expression levels of both miR-133 and miR-1 in the left ventricle. They then described that in vitro overexpression of miR-133 or miR-1 inhibited cardiac hypertrophy. In contrast, suppression of miR-133 induced hypertrophy, which was more pronounced than that after stimulation with conventional inducers of hypertrophy. In vivo inhibition of miR-133 by a single infusion of an antimiRNA antisense oligonucleotide (AMO) against miR-133 caused marked and sustained cardiac hypertrophy. They then identified specific targets of miR-133: RhoA, a guanosine diphosphate–guanosine triphosphate exchange protein regulating cardiac hypertrophy; Cdc42, a signal transduction kinase implicated in hypertrophy; and Nelf-A/WHSC2, a nuclear factor involved in cardiogenesis. Cheng et al. [41] identified 19 deregulated miRNAs in hypertrophic mouse hearts after aortic banding. Knockdown of miR-21 expression via AMO-mediated depletion had a significant negative effect on cardiomyocyte hypertrophy induced by TAC in mice or by angiotensin II or phenylephrine in cultured neonatal cardiomyocytes. Consistently, another independent group identified 17 miRNAs upregulated and three miRNAs downregulated in TAC mice and seven upregulated and four downregulated in phenylephrine-induced hypertrophy of neonatal cardiomyocytes. They further showed that inhibition of endogenous miR-21 or miR-18b that are most robustly upregulated augments hypertrophic growth, while introduction of either of these two miRNAs into cardiomyocytes represses cardiomyocyte hypertrophy [42]. A study directed to the human heart identified 67 significantly upregulated miRNAs and 43 significantly downregulated miRNAs in failing left ventricles versus normal hearts [43]. Interestingly, 86.6% of induced miRNAs and 83.7% of repressed miRNAs were regulated in the same direction in fetal and failing heart tissue compared with healthy hearts, consistent with the activation of “fetal” cardiac genes in heart failure. Bioinformatics analysis revealed that the mRNAs upregulated in the failing heart contain the putative binding sites for the downregulated miRNAs and vice versa. Most strikingly, transfection of cardiomyocytes with a set of fetal miRNAs induced cellular hypertrophy as well as changes in gene expression comparable to the failing heart. The above findings are summarized in Fig. 1.

Diagram depicting the miRNAs, along with their target genes, which have been experimentally evidenced for their participation in the development of cardiac hypertrophy. THRAP1 thyroid hormone receptor associated protein 1; RasGAP Ras GTPase-activating protein; Cdk9 cyclin-dependent kinase 9; Rheb Ras homolog enriched in brain; RhoA, a GDP–GTP exchange protein regulating cardiac hypertrophy; Cdc42, a signal transduction kinase; and Nelf-A/WHSC2, a nuclear factor involved in cardiogenesis. These proteins have been implicated in hypertrophy
It should be noted that controversies exist among these studies. Probably the most striking disparity is the contradictory results from the studies reported by Cheng et al. [41] and Tatsuguchi et al. [42]. Cheng et al. showed that knockdown of miR-21 relieves cardiomyocyte hypertrophy [41], whereas the study from Wang's [42] group demonstrated the opposite. Both studies used the phenylephrine model of hypertrophy in cultured neonatal rat ventricular myocytes. The study by Carè et al. [40] clearly indicates that miR-133 is an antihypertrophic factor and downregulation of miR-133 alone is sufficient to induce cardiac hypertrophy. However, the study reported by van Rooij et al. [38] suggests that miR-133 does not cause any morphological changes of cardiomyocytes indicative of hypertrophic growth. Moreover, among the seven studies, two [38, 40] reported downregulation of miR-133 in hypertrophy, three [39, 41, 43] failed to observe this change, one found it upregulated [42], and another study did not deal with miR-133 [32]. While miR-1 was found to be downregulated in two studies [39, 40], this observation was not reproduced in three other studies [38, 41, 42] and one study performed with human heart actually claimed it to be upregulated [43]. Increase in miR-18b was seen only in one study [42]. The most consistent changes reported by the six studies are upregulation of miR-21 (six of six studies), miR-23a (four of six), miR-125b (five of six), and miR-214 (four of six) and downregulation of miR-150 (five of six studies) and miR-30 (five of six). The discrepancies could be explained by multiple factors, including different models of hypertrophy, different time points of the same models, different miRNA microarrays for analyses, etc.
Collectively, with respect to hypertrophy, it is evident that in addition to the muscle-specific, miRNAs miR-1, miR-133, and miR-208, other miRNAs, including miR-195, miR-21, miR-18b, etc, also play an important role. It appears that multiple miRNAs are involved in cardiac hypertrophy and each of them can independently determine the pathological process.
miRNAs and cardiac arrhythmias
Arrhythmias are electrical disturbances that can result in irregular heart beating with consequent insufficient pumping of blood. Arrhythmias are often lethal, constituting a major cause for cardiac death in myocardial infarction and heart failure and being one of the most difficult clinical problems [44]. Arrhythmias can occur when there is abnormality in the electrical activities: cardiac conduction, repolarization, or automaticity. The electrical activities of the heart are determined by ion channels, the transmembrane proteins embedded across the cytoplasmic membrane of cardiomyocytes. Sodium channels and connexin43 (Cx43) are responsible for excitation generation and intercell conduction of excitations, respectively. Calcium channels account for excitation–contraction coupling and also contribute to pacemaker activities. Potassium channels govern the membrane potential and rate of membrane repolarization. Pacemaker channels, which carry the nonselective cation currents, are critical in generating sinus rhythm and ectopic heart beats as well. Intricate interplays of these ion channels maintain the normal heart rhythm. Dysfunction of any of the ion channels can render arrhythmias (Fig. 2).

Schematic illustration of the role of the muscle-specific miRNAs miR-1 and miR-133 in arrhythmias. Cx43 connexin-43; IK1 inward rectifier K+ current; IKr rapid delayed rectifier K+ current; IKs slow delayed rectifier K+ current; If pacemaker nonselective cation current or funny current; Kir2.1 a pore-forming K+ channel α-subunit for IK1; HERG a pore-forming K+ channel α-subunit for IKr; KCNQ1 a pore-forming K+ channel α-subunit for IKs; KCNE1 an auxiliary β-subunit for IKs; HCN2 and HCN4 α-subunits for If; APD action potential duration; ERP effective refractory period; EAD early after depolarization
We conducted bioinformatics analysis using miRNA database websites miRBase and miRANDA to predict the potential target genes encoding cardiac ion channel proteins for the muscle-specific miRNAs miR-1, miR-133, and miR-208. Based on the information, we then carried out luciferase reporter activity assays to verify the candidate targets. From these studies, we are able to obtain an overall picture of expression regulation of ion channel genes by the muscle-specific miRNAs and to come up with the following notes. First, while miR-1 and miR-133 both can regulate multiple genes that cover nearly all cardiac ion channel subunits, miR-208 does not seem to have any ion channel genes as its target for posttranscriptional repression. Second, miR-1 or miR-133 each has its distinct set of ion channel targets without overlapping genes for both, except for the pacemaker channel gene HCN2 that is regulated by both miR-1 and miR-133 (unpublished observations). Third, some computationally predicted targets failed to be experimentally verified but some nonpredicted genes were found to be targets for miR-1 or miR-133 according to our experimental results.
It has been commonly accepted that regional dispersion of ventricular repolarization is a marker of arrhythmogenicity risk [45, 46]. The spatial heterogeneity of cardiac repolarization is largely due to diversity and varying densities of repolarizing K+ currents [47–49]. Slowly activating delayed rectifier K+ current (I Ks) along with its underlying channel proteins KCNQ1 (pore-forming α-subunit) and KCNE1 (auxiliary β-subunit) demonstrates important spatial heterogeneity of distribution, contributing importantly to arrhythmogenicity [50]. We have experimentally established KCNQ1 and KCNE1 as targets for repression by miR-133 and miR-1, respectively (Fig. 2) [51]. More importantly, we found that the distribution of miR-133 and miR-1 transcripts within the heart is also spatially heterogeneous with the patterns corresponding to the spatial distribution of KCNQ1 and KCNE1 proteins and I Ks.
Abnormal QT interval prolongation is a prominent electrical disorder and has been proposed a predictor of mortality in patients with diabetes mellitus, presumably because it is associated with an increased risk of sudden cardiac death consequent to lethal ventricular arrhythmias [52, 53]. We have previously found that the ether-a-go-go-related gene (ERG), a long QT syndrome gene encoding a key K+ channel (I Kr) in cardiac cells, is severely depressed in its expression at the protein level but not at the mRNA level in diabetic subjects [54]. In an effort to understand the mechanisms underlying the disparate alterations of ERG protein and mRNA, we performed a study on expression regulation of ERG by miRNAs in a rabbit model of diabetes [55]. We found a remarkable overexpression of miR-133 in diabetic hearts and, in parallel, the expression of SRF, which is known to be a transactivator of miR-133, was also found robustly increased. Delivery of exogenous miR-133 into the rabbit myocytes and cell lines produced posttranscriptional repression of ERG, downregulating ERG protein level without altering its transcript level. Correspondingly, forced expression of miR-133 also caused substantial depression of I Kr, an effect abrogated by the miR-133 antisense inhibitor. Functional inhibition or gene silencing of SRF downregulated miR-133 expression and increased I Kr density. Repression of ERG by miR-133 likely underlies the differential changes of ERG protein and transcript thereby depression of I Kr and contributes to repolarization slowing thereby QT prolongation and the associated arrhythmias in diabetic hearts (Fig. 2).
One of the most deleterious alterations during myocardial infarction is the occurrence of ischemic arrhythmias [56]. We found that miR-1 is overexpressed (~2.8-fold increase) in the myocardium of individuals with coronary artery disease relative to healthy hearts. To explore the mechanisms, we used a rat model of myocardial infarction induced by occlusion of the left anterior descending coronary artery for 12 h that corresponds to the peri-infarction period during which phase II ischemic arrhythmias often occur, which represents a major challenge to our understanding and management of the disorder [57]. We found a similar increase (~2.6-fold) in miR-1 expression in the ischemic hearts of rat, which was accompanied by exacerbated arrhythmogenesis [58]. Elimination of miR-1 by an antisense inhibitor in infarcted rat hearts relieved arrhythmogenesis. miR-1 overexpression slowed cardiac conduction and depolarized the cytoplasmic membrane, which is likely the cellular mechanism for the arrhythmogenic potential of miR-1. We further established GJA1, which encodes connexin 43 [59], and KCNJ2, which encodes the K+ channel subunit Kir2.1 [60, 61], as target genes for miR-1. Cx43 is critical for intercell conductance and Kir2.1 for setting and maintaining membrane potential. Repression of these proteins by miR-1 explains for miR-1-induced slowing of cardiac conduction. We therefore proposed that myocardial infarction upregulates miR-1 expression via some unknown factors, which induces posttranscriptional repression of GJA1 and KCNJ2, resulting in conduction slowing leading to ischemic arrhythmias (Fig. 2).
Zhao et al. [29] determined in vivo miR-1-2 targets, including the cardiac transcription factor, Irx5, which represses KCND2, a potassium channel subunit (Kv4.2) responsible for transient outward K+ current (I to) [62, 63]. Their study suggests that the combined loss of Irx5 and Irx4 disrupted ventricular repolarization with a predisposition to arrhythmias. The increase in Irx5 and Irx4 protein levels in miR-1-2 mutants corresponded well with a decrease in KCND2 expression. Clearly, loss-of-function of miR-1 and Dicer mutant embryos affect conductivity through K+ channels which supports a central role for miR-1 for fine tuning the regulation of cardiac electrophysiology in pathological and normal conditions.
miRNAs and vascular angiogenesis
It is known that proliferative vascular diseases share similar cellular events and molecular mechanisms with cancer, and neointimal lesion formation is the pathological basis of proliferative vascular diseases. Neointimal growth is the balance between proliferation and apoptosis of vascular smooth muscle cells (VSMCs). The increased VSMC proliferation or the relative decreased VSMC apoptosis are responsible for neointimal lesion formation. Ji et al. [25] reported that multiple miRNAs are aberrantly expressed in the vascular wall after angioplasty with the time course changes matching the complex process of neointimal lesion formation, in which multiple genes are accordingly deregulated, as assessed with miRNA microarray. Seven days after balloon injury, 113 of the 140 artery miRNAs were differentially expressed (60 miRNAs were upregulated, and 53 miRNAs were downregulated). At 14 days after injury, 110 of the 140 artery miRNAs were differentially expressed (63 up and 47 down), whereas 102 of the 140 artery miRNAs were differentially expressed (55 up and 47 down) at 28 days after angioplasty. Their results indicate that multiple miRNAs are involved in neointimal lesion formation, but miR-21 may be of particular significance. miR-21 was found to be one of the most upregulated miRNAs in the vascular wall of balloon-injured rat carotid arteries. Inhibition of miR-21 expression via AMO-mediated miRNA depletion significantly decreased neointimal formation after angioplasty by decreasing cell proliferation and increasing cell apoptosis. Their data further identified phosphatase and tensin homolog and Bcl-2 as target genes for miR-21 in VSMCs. The authors believe that miRNAs may be a new therapeutic target for proliferative vascular diseases such as atherosclerosis, postangioplasty restenosis, transplantation arteriopathy, and stroke [25].
A study by Suárez et al. [64] showed that knockdown of Dicer by RNA interference (RNAi) techniques in human endothelial cells (ECs) altered the expression of several key regulators of endothelial biology and angiogenesis, including TEK–Tie-2, kinase insert domain receptor–vascular endothelial growth factor receptor 2, Tie-1, endothelial nitric oxide synthase (eNOS), and interleukin-8. The RNAi-mediated knockdown of Dicer in ECs resulted in a significant reduction but not complete loss of mature miRNAs. The net phenotype of Dicer knockdown in ECs was an increase in eNOS protein levels and NO release and decrease in EC growth. Forced expression of miR-222 and miR-221 that were among the highest expressed in ECs regulates eNOS protein levels after Dicer silencing.
The laboratory of Dimmeler et al. [65] also investigated the role of Dicer and Drosha in angiogenesis. In their study, ECs were transfected with small interfering RNA (siRNA) against Dicer and Drosha to inhibit miRNA biogenesis. Genetic silencing of Dicer and Drosha significantly reduced capillary sprouting of ECs and tube-forming activity. Migration of ECs was significantly decreased in Dicer siRNA-transfected cells, whereas Drosha siRNA had no effect. Silencing of Dicer but not of Drosha reduced angiogenesis in vivo. They then screened 168 human miRNAs using real-time polymerase chain reaction and revealed that members of the let-7 family, miR-21, miR-126, miR-221, and miR-222 are highly expressed in endothelial cells. Dicer and Drosha siRNA reduced lef-7f and miR-27b expression. Inhibitors against let-7f and miR-27b also reduced sprout formation indicating that these miRNAs promote angiogenesis by targeting antiangiogenic genes. In silico analysis of predicted targets for let-7 cluster identified the endogenous angiogenesis inhibitor thrombospondin-1. They confirmed that Dicer and Drosha siRNA significantly increased the expression of thrombospondin-1. Their findings indicate that inhibition of Dicer impairs angiogenesis in vitro and in vivo, whereas inhibition of Drosha induces only a minor antiangiogenic effect [65].
miRNAs and cardiomyocyte apoptosis
Loss of cardiac myocytes due to apoptotic cell death is common to myocardial ischemia, cardiac hypertrophy, and heart failure. Increasing evidence indicates that miRNAs as apoptosis regulators are also implicated cardiovascular disease.
We have recently shown that miR-1 level is markedly elevated in ischemic myocardium where apoptotic cell death plays an important role in the detrimental changes of the diseased heart [58]. Coincidently, another study from our laboratories revealed miR-1 and miR-133 produced opposing effects on apoptosis induced by oxidative stress in H9c2 rat ventricular cells, with miR-1 being proapoptotic and miR-133 being antiapoptotic [66]. miR-1 level was significantly increased in response to oxidative stress and this increase reduced the protein levels of the antiapoptotic molecules heat shock protein-60 (HSP60) [67] and HSP70 [68] without changing their transcript levels. By comparison, miR-133 repressed apoptotic caspase-9 [69] expression at both protein and mRNA levels to favor cell survival. The posttranscriptional repression of HSP60–HSP70 and caspase-9 was further confirmed by luciferase reporter experiments [66]. Our results therefore suggest that the ischemic elevation of miR-1 level may contribute to the enhanced apoptotic cell death in myocardial infarction, in addition to the enhanced arrhythmogenicity. In another case, we found that in the heart with cardiomyopathy of diabetic subjects, miR-133 level is increased [55]. It is speculated that this increase may serve to protect the heart considering that miR-133 negatively regulate apoptosis.
Also noteworthy is that among the hypertrophy-inducing miRNAs, miR-1, miR-133, and miR-21 are known apoptosis regulators. Other miRNAs miR-195, miR-18b, and miR-208 that have been shown to independently regulate hypertrophy also have the potential to regulate apoptosis, although experimental data are still missing. According to our bioinformatics prediction, miR-195 could repress multiple survival genes including the Bcl-2 gene family, the E2F gene family, Akt, and NF2, being characteristic of a proapoptotic regulator. Similarly, miR-18b also has the potential to regulate multiple antiapoptotic genes, including Akt, Sp1, Bcl-2, E2F, and IGF1.
miRNA interference technologies and cardiovascular disease
The role of miRNAs in the pathogenesis of the heart and vessel points to a possibility of miRNAs as targets for treatment of cardiovascular disease. To achieve the therapeutic goal, strategies for manipulating miRNAs by influencing their expression, stability, and function are needed. Such miRNA interference strategies involve both “gain-of-function” and “loss-of-function” approaches. Several innovative technologies have been developed for this purpose over the past few years.
Supply of artificial miRNAs
Some miRNAs are decreased in their expression levels in cardiovascular tissues and correction of deregulated expression of these miRNAs by “replacement therapy” may be an efficient approach for management of the disorders. Introduction of synthetic miRNAs into cells as a “gain-of-function” approach has proven feasible under many conditions.
AntimiRNA antisense inhibitor oligonucleotides techniques
AMOs are artificially designed oligonucleotides fully complementary to target miRNAs and it acts to breakdown the targeted miRNAs with unknown mechanisms [51, 55, 58, 70, 71]. In this way, AMO abolishes the action of the targeted miRNAs. It mediates potent and miRNA-specific inhibition of miRNA function, providing a powerful “loss-of-function” strategy for interfering miRNA expression. When attached with cholesterol moieties to permit entry into the cell, an AMO is called “antagomirs”. Antagomirs can be used to bind with unwanted miRNAs and have been shown to be capable of in vivo silencing expression of miRNAs in the mouse heart [72]. Today, AMO has become a necessary tool for miRNA research and has also been proposed to be potential therapeutic agents as well.
miRNA sponge techniques
Ebert et al. [73] invented an innovative antimiRNA approach termed “miRNA sponges.” The idea behind it is to produce a single specie of RNAs containing multiple, tandem-binding sites for a miRNA seed family of interest, in order to target all members of that miRNA seed family, taking advantage of the fact that the interaction between miRNA and target is nucleated by and largely dependent on base pairing in the seed region (positions 2–8 of the miRNA). The authors constructed sponges by inserting tandemly arrayed miRNA binding sites into the 3'UTR of a reporter gene encoding destabilized green fluorescent protein driven by the cytomegalovirus promoter, which can yield abundant expression of the competitive inhibitor transcripts. Binding sites for a particular miRNA seed family were perfectly complementary in the seed region with a bulge at positions 9–12 to prevent RNA-interference-type cleavage and degradation of the sponge RNA. When vectors encoding these miRNA sponges are transiently transfected into cultured cells, they depress miRNA targets as strongly as the conventional AMOs. The major advancement of this technique over the AMO technique is that it can better inhibit functional classes of miRNAs than do AMOs that are designed to block single miRNA sequences.
miRNA-masking antisense oligonucleotides techniques
It has been predicted that each single miRNA may regulate as many as 1,000 protein-coding genes and each gene may be regulated by multiple miRNAs [23]. This implies that the action of miRNAs is binding sequence specific but not gene specific; similarly, the action of AMOs is miRNA specific but not gene specific either. These properties of miRNAs and AMOs may present the obstacles for development as therapeutic agents because they may elicit unwanted side effects and toxicity due to their non-gene-specific functional profiles. To tackle this problem, we have developed a technique that we term miRNA-masking antisense oligodeoxynucleotides (miR-Mask ODN) [74]. We validated this technology by testing its application to the cardiac pacemaker channel-encoding genes HCN2 and HCN4. A miR-Mask is designed to be exactly antisense to the binding site for an endogenous miRNA of interest in 3'UTR of the target mRNA. Once introduced into the cell, the miR-Mask can form duplex with the target mRNA with higher affinity than that endogenous miRNA. In this way, the miR-Mask acts as a target protector masking the binding site to block the access of that miRNA to its site of action with consequent relief of the repressive effect produced by that miRNA. We have successfully created miR-Masks that can bind to HCN2 and HCN4 and prevent the repressive actions of miR-1 and miR-133. These miR-Masks result in enhanced protein expression of the pacemaker channels and increased pacemaker activities revealed by whole-cell patch-clamp recordings. Functionally, the miR-Masks for HCN channels cause acceleration of heart rate in rats, simulating “biological pacemakers” [75]. The major advantage of this technology is that it offers a gene-specific miRNA-interfering strategy. This technology has been validated by a recent study in which the authors investigated the role of zebrafish miR-430 in regulating expression of TGF-β nodal agonist squint and antagonist lefty [76], the key regulators of mesendoderm induction and left–right axis formation. They designed target protector morpholinos complementary to miRNA binding sites in target mRNAs in order to disrupt the interaction of specific miRNA–mRNA pairs. Protection of squint or lefty mRNAs from miR-430 resulted in enhanced or reduced nodal signaling, respectively.
The miR-Mask approach defers from the AMO approach in several aspects, despite that they both can result in enhancement of gene expression by removing the repressive effects of a particular miRNA on protein translation of the target mRNA. First, an AMO is designed to entirely base pair with the sequence of the target miRNA, whereas a miR-Mask is designed based on the sequence of the target site for a miRNA in the 3′UTR of the target mRNA [74]. Second, an AMO interacts with (binds to) its target miRNA and may well cause degradation of that miRNA such that all functions of that miRNA are deemed to be impaired, whereas a miR-Mask interacts with (binds to) its target mRNA and does not induce miRNA degradation such that the function of that miRNA on other genes is intact. In this sense, a miR-Mask is not only a target protector but also a miRNA protector. Third, the action of an AMO is miRNA specific but not gene specific and it may well induce enhancement of expression of multiple genes regulated by the target miRNA, whereas a miRNA-masking antisense ODN is expected to be gene specific because it is fully complementary to the target mRNA sequence and is miRNA specific as well because it is designed to target the binding site of that miRNA. Based on the above discussions, it seems that the miR-Mask approach is a valuable supplement to the AMO technique; while AMO is indispensable for studying the overall function of a miRNA, the miR-Mask might be more appropriate for studying the specific outcome of regulation of the target gene by the miRNA. On the other hand, the miR-Mask approach defers from the conventional antisense technique in the following two aspects, despite that they both are entirely complementary to the target sequences. First, a conventional antisense ODNs can be designed to target any part of the protein-coding region of a gene (though the sequences from the translation start codon are frequently used), whereas a miR-Mask are limited to the target site of a miRNA in the 3′UTR of a protein-coding gene. In other words, the action of a miR-Mask depends on the basal activity of the endogenous miRNA on the target mRNA. Second, more importantly, a conventional antisense ODNs binds to its target site in the coding region of a gene and hinder the protein translation process, whereas a miR-Mask bind to the 3′UTR and masks the target site of a miRNA so as to block the action of the endogenous miRNA and enhance protein translation. Thus, the two techniques produce exactly opposite outcomes: one inhibits but the other enhances gene expression. This expression-enhancing action of miR-Mask is unique and could have many applications. A comparison of the miR-Mask, the AMO, and the conventional antisense ODN techniques are summarized in Table 1 and illustrated in Fig. 3.

Upper panel: Schematic presentation of actions of miRNA-masking antisense oligonucleotide (miR-Mask) compared with the conventional antisense oligodeoxynucleotide (ODN) and antimiRNA antisense inhibitor oligonucleotide (AMO) technologies. Synthetic nucleic acids are introduced into the cells. Antisense ODNs bind to the coding region of the target mRNA and hinder the translation process; AMOs bind to the target miRNA, resulting in miRNA cleavage; miR-Masks bind to the binding site of miRNAs in 3′UTR of the target mRNA and prevent miRNAs from binding to the target mRNA, leading to a relief of translational repression without affecting miRNAs. Lower panel: Schematic presentation of actions of miRNA mimic (miR-Mimic) compared with the miRNA and small interference RNA (siRNA). Synthetic miR-Mimic and siRNA are introduced into the cells and endogenous miRNA is synthesized by the cell. siRNAs bind to the coding region of target miRNAs and cause mRNA cleavage; miRNAs bind to 3′UTR of multiple target mRNAs and produce non-gene-specific posttranscriptional repression to inhibit translation; miR-Mimics bind to 3′UTR of unique target mRNAs and produce gene-specific posttranscriptional repression to inhibit translation
miRNA mimic techniques (miR-Mimic)
With a rationale similar to miR-Mask, in order to circumvent the problem of non-gene specificity of miRNA actions, we developed a technology utilizing synthetic, unnatural nucleic acids that can bind to the unique sequence of target mRNAs in a gene-specific manner and elicit posttranscriptional regulatory effects as a miRNA does [74]. We name this miRNA Mimic technology (miR-Mimic). Two conditions must be satisfied in order to generate a miRNA mimic: (1) a miR-Mimic must be complementary to, only to, the 3′UTR of the target gene to elicit miRNA action, so that that it represses the target gene at the posttranscriptional level with minimal effects on the mRNA level; and (2) the 3′UTR of the target gene must contain a unique sequence distinct from other genes to elicit gene-specific action. We examine the application of the technique to cardiac pacemaker genes HCN2 and HCN4. Following the rules, we first identified a stretch of sequence in the 3′UTR unique to the HCN2 (or HCN4) gene that is expectedly long enough for miRNA action. Based on the unique sequence, we designed a 22-nt miR-Mimic that at the 5′ end has eight nucleotides (nucleotides 2–8) and at the 3′end has seven nucleotides, complementary to the HCN2 (or HCN4) sequence. These miR-Mimics produced substantial repression of HCN channel protein expression with concomitant depression of pacemaker activities and reduction of heart rate but with minimal effects on their mRNA levels. We have also tested the miR-Mimics designed to target Bcl-2 and E2F, respectively, and observed remarkable suppression of tumor cell growth (unpublished data). The results demonstrated a promise of utilizing the technology for gene-specific repression of expression at the protein level based on the principle of miRNA actions. The differences among miR-Mimics and natural miRNAs and siRNAs are listed in Table 2 and illustrated in Fig. 3.
References
Latronico MVG, Catalucci D, Condorelli G (2007) Emerging role of microRNAs in cardiovascular biology. Circ Res 101:1225–1236
Callis TE, Chen JF, Wang DZ (2007) MicroRNAs in skeletal and cardiac muscle development. DNA Cell Biol 26:219–225
van Rooij E, Olson EN (2007) MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest 117:2369–2376
Chien KR (2007) MicroRNAs and the tell-tale heart. Nature 447:389–390
Anderson ME, Mohler PJ (2007) Micro RNA may have macro effect on sudden death. Nat Med 13:410–411
Mishima Y, Stahlhut C, Giraldez AJ (2007) miR-1-2 gets to the heart of the matter. Cell 129:248–249
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23:4051–4060
Lee Y, Jeon K, Lee JT, Kim S, Kim VN (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21:4663–4670
Kim VN (2004) MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol 14:156–159
Peters L, Meister G (2007) Argonaute proteins: mediators of RNA silencing. Mol Cell 26:611–623
Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R (2005) MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol 7:719–723
Brennecke J, Stark A, Russell RB, Cohen SM (2005) Principles of microRNA-target recognition. PLoS Biol 3:404–418
Jackson RJ, Standart N (2007) How do microRNAs regulate gene expression? Sci STKE 23:243–249
Lewis BP, Shih I-H, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115:787–798
Nilsen TW (2007) Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet 23:243–249
Pillai RS, Bhattacharyya SN, Filipowicz W (2007) Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol 17:118–126
Zhao Y, Samal E, Srivastava D (2005) Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 436:214–220
Vasudevan S, Tong Y, Steitz JA (2007) Switching from repression to activation: microRNAs can up-regulate translation. Science 318:1931–1934
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM (2005) Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433:769–773
Alvarez-Garcia I, Miska EA (2005) MicroRNA functions in animal development and human disease. Development 132:4653–4662
Ambros V (2004) The functions of animal microRNAs. Nature 431:350–355
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20
Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I (2006) A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 126:1203–1217
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T (2002) Identification of tissue-specific miRNAs from mouse. Curr Biol 12:735–739
Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C (2007) MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res 100:1579–1588
Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ (2006) The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38:228–233
Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF (2006) Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci U S A 103:8721–8726
Kwon C, Han Z, Olson EN, Srivastava D (2005) MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci U S A 102:18986–18991
Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D (2007) Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129:303–317
Sokol NS, Ambros V (2005) Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev 19:2343–2354
Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH (2005) MicroRNA expression in zebrafish embryonic development. Science 309:310–311
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN (2007) Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316:575–579
Niu Z, Li A, Zhang SX, Schwartz RJ (2007) (2007) Serum response factor micromanaging cardiogenesis. Curr Opin Cell Biol 19:918–627
Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, Richardson JA, Bassel-Duby R, Olson EN (2007) An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc Natl Acad Sci U S A 104:20844–20849
Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF (2005) MicroRNAs regulate brain morphogenesis in zebrafish. Science 308:833–838
Srivastava D, Thomas T, Lin Q, Kirby ML, Brown D, Olson EN (1997) Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat Genet 16:154–160
McKinsey TA, Olson EN (2005) Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J Clin Invest 115:538–546
van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN (2006) A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A 103:18255–18260
Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M (2007) MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100:416–424
Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med 13:613–618
Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C (2007) MicroRNAs are aberrantly expressed in hypertrophic heart. Do they play a role in cardiac hypertrophy? Am J Pathol 170:1831–1840
Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ (2007) Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 42:1137–1141
Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J (2007) MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116:258–267
Akar FG, Tomaselli GF (2005) Ion channels as novel therapeutic targets in heart failure. Ann Med 37:44–54
Day CP, McComb JM, Campbell RWF (1990) QT dispersion: an indication of arrhythmia risk in patients with long QT intervals. Br Heart J 63:342–344
Roden DM, Yang T (2005) Protecting the heart against arrhythmias: potassium current physiology and repolarization reserve. Circulation 112:1376–1378
Liu DW, Antzelevitch C (1995) Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res 76:351–365
Verduyn SC, Vos MA, van der Zande J, van der Hulst FF, Wellens HJ (1997) Role of interventricular dispersion of repolarization in acquired torsade-de-pointes arrhythmias: Reversal by magnesium. Cardiovasc Res 34:453–463
Szentadrassy N, Banyasz T, Biro T, Szabo G, Toth BI, Magyar J, Lazar J, Varro A, Kovacs L, Nanasi PP (2005) Apico-basal inhomogeneity in distribution of ion channels in canine and human ventricular myocardium. Cardiovasc Res 65:851–860
Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P, Nagy Z, Bogats G, Lathrop DA, Papp JG, Varro A (2005) Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 112:1392–1399
Luo X, Lin H, Lu Y, Li B, Xiao J, Yang B, Wang Z (2007) Transcriptional activation by stimulating protein 1 and post-transcriptional repression by muscle-specific microRNAs of IKs-encoding genes and potential implications in regional heterogeneity of their expressions. J Cell Physiol 212:358–367
Cardoso C, Salles G, Bloch K, Deccache W, Siqueira-Filho AG (2001) Clinical determinants of increased QT dispersion in patients with diabetes mellitus. Int J Cardiol 79:253–262
Veglio M, Chinaglia A, Cavallo-Perin P (2004) QT interval, cardiovascular risk factors and risk of death in diabetes. J Endocrinol Invest 27:175–181
Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, Zhang H, Bai Y, Yang B, Wang Z (2006) Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of the abnormal QT prolongation and the associated arrhythmias in diabetic rabbits. Am J Physiol 291:1446–1455
Xiao J, Luo X, Lin H, Xu C, Gao H, Wang H, Yang B, Wang Z (2007) MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem 282:12363–12367
Ghuran AV, Camm AJ (2001) Ischaemic heart disease presenting as arrhythmias. Br Med Bull 59:193–210
Clements-Jewery H, Hearse DJ, Curtis MJ (2005) Phase 2 ventricular arrhythmias in acute myocardial infarction: a neglected target for therapeutic antiarrhythmic drug development and for safety pharmacology evaluation. Br J Pharmacol 145:551–564
Yang B, Lin H, Xiao J, Luo X, Li B, Lu Y, Wang H, Wang Z (2007) The muscle-specific microRNA miR-1 causes cardiac arrhythmias by targeting GJA1 and KCNJ2 genes. Nat Med 13:486–491
Jongsma HJ, Wilders R (2000) Gap junctions in cardiovascular disease. Circ Res 86:1193–1197
\Diaz RJ, Zobel C, Cho HC, Batthish M, Hinek A, Backx PH, Wilson GJ (2004) Selective inhibition of inward rectifier K+ channels (Kir2.1 or Kir2.2) abolishes protection by ischemic preconditioning in rabbit ventricular cardiomyocytes. Circ Res 95:325–332
Wang Z, Yue L, White M, Pelletier G, Nattel S (1998) Differential expression of inward rectifier potassium channel mRNA in human atrium versus ventricle and in normal versus failing hearts. Circulation 98:2422–2428
Wang Z, Feng J, Shi H, Pond A, Nerbonne JM, Nattel S (1999) The potential molecular basis of different physiological properties of transient outward K+ current in rabbit and human hearts. Circ Res 84:551–561
Costantini DL, Arruda EP, Agarwal P, Kim K-H, Zhu Y, Lebel M, Cheng CW, Park CY, Pierce S, Guerchicoff A, Pollevick GD, Chan TY, Kabir MG, Cheng SH, Husain M, Antzelevitch C, Srivastava D, Gross GJ, Hui CC, Backx PH, Bruneau BG (2005) The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell 123:347–358
Suárez Y, Fernández-Hernando C, Pober JS, Sessa WC (2007) Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res 100:1164–1173
Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S (2007) Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res 101:59–68
\Xu C, Lu Y, Lin H, Xiao, J, Wang H, Luo X, Li B, Yang B, Wang Z (2007) The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis via targeting HSP60/HSP70 and caspase-9 in cardiomyocytes. J Cell Sci 120:3045–3052
Kirchhoff SR, Gupta S, Knowlton AA (2002) Cytosolic HSP60, apoptosis, and myocardial injury. Circulation 105:2899–2904
Marber MS, Mestril R, Chi SH, Sayen MR (1995) Overexpression of the rat inducible 70 kDa heat shock protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest 95:1446–1456
\Han Y, Chen YS, Liu Z, Bodyak N, Rigor D, Bisping E, Pu WT, Kang PM (2006) Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ Res 99:415–423
Weiler J, Hunziker J, Hall J (2006) Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease? Gene Ther 13:496–502
Hammond SM (2006) MicroRNA therapeutics: a new niche for antisense nucleic acids. TiMM 12:99–101
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M (2005) Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438:685–689
Ebert MS, Neilson JR, Sharp PA (2007) MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4:721–726
Xiao J, Yang B, Lin H, Lu Y, Luo X, Wang Z (2007) Novel approaches for gene-specific interference via manipulating actions of microRNAs: examination on the pacemaker channel genes HCN2 and HCN4. J Cell Physiol 212:285–292
Robinson RB, Brink PR, Cohen IS, Rosen MR (2006) I f and the biological pacemaker. Pharmacol Res 53:407–415
Choi WY, Giraldez AJ, Schier AF (2007) Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science 318:271–274
Acknowledgements
The authors thank XiaoFan Yang for excellent technical support. This work was supported in part by the Canadian Institute of Health Research, Fonds de la Recherche de l'Institut de Cardiologie de Montreal, awarded to Dr. Z Wang, and by the National Basic Research Program of China (973 Program; 2007CB512000/2007CB512006) awarded to Dr. B Yang. Dr. Z Wang is a senior research scholar of the Fonds de Recherche en Sante de Quebec, a LongJiang Scholar Professor, China, and a ChangJiang Scholar Professor, China.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wang, Z., Luo, X., Lu, Y. et al. miRNAs at the heart of the matter. J Mol Med 86, 771–783 (2008). https://doi.org/10.1007/s00109-008-0341-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-008-0341-3