
Abstract
A series of arylpiperazine derivatives of 1,16-diphenyl-19-azahexacyclo-[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione and 4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione was synthesized. The pharmacological profile of compound 4 at the 5-HT1A receptor was measured by binding assay. The title compounds were tested in cell-based assay against the human immunodeficiency virus type-1. The X-ray crystallographic studies of derivatives 2, 6, 7, 11, 19, and 20 were presented.
Similar content being viewed by others

Introduction
The literature survey shows that many ligands of serotonin 5-HT1A, 5-HT2A, and 5-HT7 receptors contain a flexible hydrocarbon chain of different lengths, attached to an arylpiperazine moiety that is the pharmacophore group (Fig. 1) (Lewgowd et al., 2011; Czopek et al., 2010; Bojarski, 2006; Leopoldo, 2004). The pharmacophore group is recognized not only by metabotropic serotonin receptor binding sites, but also by those of D2-dopaminergic (González-Gómez et al., 2003) and α1-adrenergic receptors (Prandi et al., 2012).

Some representative 5-HT1A receptor ligands
Using quantitative structure–activity relationship analysis, the “rule of five” scheme was worked out for orally administrated drugs (Lipinski et al., 1997; Kerns and Di, 2008). According to authors, the drugs that cross the blood–brain barrier are those of molecular mass lower than 450 u and of theoretical partition coefficient n-octanol/water (logP) being in the range of 1–4 or logD 7.4 1–3. The biological barrier permeability is also determined by the following important parameters: numbers of hydrogen bond donors and acceptors in the potential medicine’s structure (HBD maximum 4 and HBA less than 6), polar surface area (PSA) correlated with them [expected value is less than 60–70 Å2 (Oprea, 2002)], as well as compound’s solubility (logS greater than 60 μg/cm3). Proper drug permeability makes it possible to cross the barrier and to reach the regions of a drug’s action.
In last two decades, a number of binding modes of long-chain arylpiperazine derivatives to 5-HT1A (Lewgowd et al., 2011; Nowak et al., 2006), 5-HT2A (Klabunde and Evers, 2005; Bronowska et al., 2001), and 5-HT7 (Kim et al., 2012; López-Rodríguez et al., 2003) receptors have been proposed. The ionic interaction between the protonated nitrogen of the piperazine ring of a ligand and Asp3.32 residue of the receptor (Nowak et al., 2006; Vermeulen et al., 2003; Roth et al., 1997) constituted a main essential interaction. The hydrophobic terminal imide or amide group, the hydrocarbon linker, and an aromatic ring bound to the piperazine moiety are placed in a hydrophobic pocket composed of aromatic and/or aliphatic amino acids side chains (Kim et al., 2012; Varin et al., 2010; Lepailleur et al., 2005). The flexible chain of N-(4-arylpiperazin-1-yl-alkyl)substituted derivatives can adopt one of the two main conformations: extended or bent. The results of geometry optimization (Lewgowd et al., 2011) proved that conformers with extended spacer are preferred in a solution, whereas in vacuum bent geometries predominate. Theoretical calculations determine minimum energy for extended linker conformations also in solid state and for complexes with a receptor (Siracusa et al., 2008). According to pharmacophore model of the 5-HT1A receptor (Chilmonczyk et al., 1997; Bronowska et al., 2001), a folded conformer promotes high affinity for the 5-HT1A receptor. It is known that ligand binding can lead to a change in the conformation of the receptor protein, however, also in the ligand itself (Sylte et al., 2001). In addition, the role of the solvent molecules is quite difficult to explain—they can take part in a ligand—receptor H-bond formation, be involved in the process of a receptor activation or influence entropy effects (Pardo et al., 2007).
This paper reports synthesis and biological activity of compounds purposely designed to combine the bulky hydrophobic imide ring with alkyl linker bearing different substituents. The collected group of arylpiperazine derivatives can be used for further investigations concerning ligand-5-HT receptor interactions. For this reason X-ray crystallographic studies of derivatives 2, 6, 7, 11, 19, and 20 were described. The molecular descriptors for selected arylpiperazine derivatives were presented. The pharmacological profile of the compound 4 was evaluated for its affinity to the 5-HT1A receptor. It was reported, that cytotoxicity of aromatic, high-volume arylpiperazine derivatives is low (Filosa et al., 2007; Ananda Kumar et al., 2009), and they act as anti-HIV-1 agents (Yang et al., 2010), cytotoxicity and anti-HIV activity of selected derivatives were examined.
Materials and methods
Chemistry
All chemicals and solvents were purchased from Aldrich. Melting points were determined on an Electrothermal Digital Melting Point Apparatus and are uncorrected. The NMR spectra were recorded on a Bruker AVANCE DMX400 spectrometer, operating at 300 MHz (1H NMR) and 75 MHz (13C NMR). The chemical shift values are expressed in ppm relative to TMS as an internal standard. Mass spectral electrospray ionization (ESI) measurements were carried out on a Mariner Perspective—Biosystem instrument with TOF detector. The spectra were obtained in the positive ion mode with a declustering potential 140–300 V. Elemental analyses were recorded on a CHN model 2400 Perkin-Elmer. TLC was carried out using silica gel 60 F254 with layer thickness of 0.25 mm (Merck) and the results were visualized using UV lamp at 254 nm. Column chromatography was carried out using silica gel 60 (200–400 mesh, Merck) and chloroform/methanol (19.5:0.5 vol) mixture as eluent.
1,16-Diphenyl-19-azahexacyclo[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (1)
The mixture of 2.14 g (0.004 mol) of 1,3-diphenyl-2H-cyclopenta[l]phenanthren-2-one (“Phencyclone”) was suspended in 75 ml of benzene and 0.48 g (0.005 mol) of maleimide was added. After refluxing time of 8 h the residue was evaporated, and the residue was purified by column chromatography (chloroform:methanol 9.5:0.5 vol). The combined fractions were condensed to dryness to give 1.86 g (87 %) of (1), m.p. 327–328 °C. 1H NMR (DMSO-d 6) δ (ppm): 11.04 (s, 1H, NH), 8.85 (d, 2H, CHarom., J = 8.4 Hz), 8.24 (d, 2H, CHarom., J = 7.8 Hz), 7.73 (t, 2H, CHarom., J = 7.2 Hz), 7.59–7.51 (m, 4H, CHarom.), 7.41 (t, 2H, CHarom., J = 7.5 Hz), 7.22 (t, 2H, CHarom., J = 7.8 Hz), 7.14 (d, 2H, CHarom., J = 7.8 Hz), 7.04 (d, 2H, CHarom., J = 5.7 Hz), 4.65 (s, 2H, CH). 13C NMR (DMSO-d 6) δ (ppm): 197.12, 173.09, 173.05, 134.03, 133.98, 133.48, 133.22, 133.76 (2C), 132.37, 132.12, 132.09, 132.06, 132.00, 131.83, 131.62, 131.47, 130.49, 130.21, 129.75, 129.68 (2C), 128.63, 128.54, 127.96, 126.84, 126.78, 122.35, 122.31, 63.65, 63.59, 45.25, 45.20. Anal. Calcd. for C33H21NO3: C, 82.45; H, 4.38; N, 2.92. Found: C, 82.40; H, 3.00; N, 4.40.
19-(4-Bromobutyl)-1,16-diphenyl-19-azahexacyclo-[14.5.1.02,15.03,8.09,14.017,21]-docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (2)
A mixture of imide (1) (1.41 g, 0.003 mol), 1,4-dibromobutane (0.7 ml, 0.006 mol), anhydrous K2CO3 (1.39 g), and catalytic amount of KI were refluxed in acetonitrile for 24 h. Then the solvent was removed on a rotary evaporator and the oily residue was purified by column chromatography (chloroform:methanol 9.5:0.5 vol). The combined fractions were condensed to dryness to give 1.36 g (86 %) of (2), m.p. 286–289 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.84 (d, 2H, CHarom., J = 9.0 Hz), 8.27 (d, 2H, CHarom., J = 8.4 Hz), 7.75 (t, 2H, CHarom., J = 8.1 Hz), 7.59–7.52 (m, 4H, CHarom.), 7.43 (t, 2H, CHarom., J = 8.7 Hz), 7.25–7.14 (m, 4H, CHarom.), 7.01 (d, 2H, CHarom., J = 7.5 Hz), 4.61 (s, 2H, CH), 2.87–2.78 (m, 2H, CH2), 2.11–2.07 (m, 2H, CH2), 1.24–1.21 (m, 2H, CH2), 0.49–0.43 (m, 2H, CH2). 13C NMR (DMSO-d 6) δ (ppm): 197.09, 173.12, 173.01, 134.11, 133.88, 133.51 (2C), 133.28, 133.39, 132.32, 132.17, 132.04, 132.00, 131.90, 131.87, 131.65, 131.36, 130.27, 130.19, 129.83, 129.69, 129.66, 128.52, 128.47, 127.89, 126.72, 126.68, 122.33, 122.30, 63.68, 63.61, 45.31, 45.28, 44.89, 32.79, 28.74, 28.53. ESI MS: m/z = 638.0 [M+H]+ (100 %).
General method for the preparation of arylpiperazine derivatives of 19-(4-bromobutyl)-1,16-diphenyl-19-azahexacyclo[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (3–9)
A mixture of derivative (2) (0.3 g, 0.002 mol) and the corresponding amine (0.004 mol), anhydrous K2CO3 (0.3 g), and catalytic amount of KI were refluxed in acetonitrile for 30 h. Then the mixture was filtered off and the solvent was evaporated. The gray residue was purified by column chromatography (chloroform:methanol 9.5:0.5 vol) and/or crystallized from methanol. Obtained compounds were converted into their hydrochlorides. The solid product was dissolved in methanol saturated with gaseous HCl. The hydrochloride was precipitated by addition of diethyl ether. The crude product was crystallized from an appropriate solvent.
1,16-Diphenyl-19-(4-(4-pyridin-2-ylpiperazin-1-yl)butyl)-19-azahexacyclo-[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (3)
Yield: 67 %, m.p. 200–203 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.81 (d, 2H, CHarom., J = 8.7 Hz), 8.27 (d, 2H, CHarom., J = 8.1 Hz), 8.09–8.06 (m, 1H, CHarom.), 7.74 (t, 2H, CHarom., J = 7.8 Hz), 7.57–7.40 (m, 7H, CHarom.), 7.36–7.14 (m, 4H, CHarom.), 7.05 (d, 2H, CHarom., J = 9.3 Hz), 6.75 (d, 1H, CHarom., J = 8.7 Hz), 6.60 (d–d, 1H, CHarom., J 1 = 5.1 Hz, J 2 = 5.4 Hz), 4.67 (s, 2H, CH), 3.78 (s, 1H, CH2), 3.31–2.72 (m, 3H, CH2), 3.05 (s, 1H, CH2), 2.92 (s, 1H, CH2), 2.05 (t, 4H, CH2, J = 2.1 Hz), 1.44 (t, 2H, CH2, J = 7.2 Hz), 1.24–1.22 (m, 1H, CH2), 0.88–0.83 (m, 1H, CH2), 0.33–0.23 (m, 2H, CH2). 13C NMR (DMSO-d 6) δ (ppm): 197.17, 173.08, 173.02, 157.48, 147.68, 137.35, 134.24, 133.73, 133.68, 133.35, 133.30, 132.12 (3C), 132.07, 132.02, 132.00, 131.87, 131.69, 131.51, 130.31, 130.12, 129.99, 129.84, 129.73, 128.47, 128.32, 127.77, 126.58, 126.49, 122.41, 122.19, 119.83, 108.92, 63.75, 63.72, 50.87, 50.43, 48.58, 48.49, 45.34, 45.32, 44.86, 32.69, 28.81, 28.73. ESI MS: m/z = 697.1 [M+H]+ (100 %).
19-(4-(4-(2-(Methyloxy)phenyl)piperazin-1-yl)butyl)-1,16-diphenyl-19-azahexa-cyclo[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (4)
Yield: 71 %, m.p. 197–200 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.83 (d, 2H, CHarom., J = 8.4 Hz), 8.27 (d, 2H, CHarom., J = 7.8 Hz), 7.74 (t, 2H, CHarom., J = 7.8 Hz), 7.58–7.52 (m, 4H, CHarom.), 7.42 (t, 2H, CHarom., J = 7.5 Hz), 7.24–7.14 (m, 4H, CHarom.), 7.10 (d, 2H, CHarom., J = 8.7 Hz), 6.92–6.83 (m, 4H, CHarom.), 4.68 (s, 2H, CH), 3.75 (s, 3H, OCH3), 2.78–2.72 (m, 7H, CH2), 2.17–2.12 (m, 4H, CH2), 1.44 (t, 3H, CH2, J = 7.2 Hz), 1.23–1.16 (m, 1H, CH2), 1.05 (t, 1H, CH2, J = 6.9 Hz). 13C NMR (DMSO-d6) δ (ppm): 197.14, 173.11, 173.09, 157.44, 147.52, 142.74, 137.31, 134.27, 133.79, 133.66, 133.31 (2C), 133.30, 132.16 (2C), 132.03, 132.01, 131.96, 131.83, 131.68, 131.57, 130.34, 130.05, 129.94, 129.81, 129.78, 128.44, 128.29, 127.68, 126.53, 126.47, 122.46, 122.21, 119.80, 108.87, 63.74, 63.71, 55.12, 50.85, 50.46, 48.53, 48.47, 45.35, 45.31, 44.88, 32.67, 28.78, 28.74. ESI MS: m/z = 726.1 [M+H]+ (100 %).
1,16-Diphenyl-19-(4-(4-phenylpiperazin-1-yl)butyl)-19-azahexacyclo-[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (5)
Yield: 69 %, m.p. 202–203 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.71 (d, 2H, CHarom., J = 8.1 Hz), 8.31 (d, 2H, CHarom., J = 8.1 Hz), 7.62–7.69 (m, 2H, CHarom.), 7.64–7.48 (m, 7H, CHarom.), 7.45–7.37 (m, 3H, CHarom.), 7.22–7.14 (m, 6H, CHarom.), 7.08–7.04 (m, 1H, CHarom.), 4.48 (s, 2H, CH), 3.51–3.42 (m, 4H, CH2), 3.27–3.23 (m, 3H, CH2), 3.13–2.95 (m, 4H, CH2), 2.63–2.61 (m, 2H, CH2), 2.35–2.29 (m, 3H, CH2). 13C NMR (DMSO-d 6) δ (ppm): 197.23, 173.17, 173.09, 157.53, 147.75, 137.42, 134.33, 133.82, 133.79, 133.41, 133.32, 132.17, 132.11, 132.06, 132.03, 131.92, 131.77 (2C), 131.58, 130.43, 130.18, 129.98, 129.89, 129.78 (2C), 128.51, 128.39, 127.81, 126.62, 126.53, 122.48, 122.22, 119.86, 115.37, 115.29, 63.81, 63.78, 50.90, 50.62, 48.64, 48.54, 45.48, 45.46, 44.93, 32.70, 28.84, 28.77. ESI MS: m/z = 696.2 [M+H]+ (100 %).
19-(4-(4-(4-Chlorophenyl)piperazin-1-yl)butyl)-1,16-diphenyl-19-azahexacyclo-[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (6)
Yield: 92 %, m.p. 220–221 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.15 (d, 2H, CHarom., J = 8.4 Hz), 8.27 (d, 2H, CHarom., J = 7.5 Hz), 7.74 (t, 2H, CHarom., J = 7.8 Hz), 7.57–7.52 (m, 4H, CHarom.), 7.42 (t, 2H, CHarom., J = 7.5 Hz), 7.24–7.13 (m, 6H, CHarom.), 7.02 (d, 2H, CHarom., J = 8.7 Hz), 6.88 (d, 2H, CHarom., J = 9.3 Hz), 4.67 (s, 2H, CH), 3.49–3.43 (m, 4H, CH2), 3.28–3.20 (m, 3H, CH2), 3.15–2.99 (m, 4H, CH2), 2.69–2.59 (m, 2H, CH2), 2.37–2.30 (m, 3H, CH2). 13C NMR (DMSO-d 6) δ (ppm): 197.21, 173.11, 173.06, 157.50, 147.74, 137.41, 134.36, 133.81, 133.78, 133.43, 133.33, 132.15, 132.12, 132.07, 132.04, 131.95, 131.72, 131.68, 131.56, 130.46, 130.12, 129.97, 129.84, 129.73 (2C), 128.59, 128.37, 127.85, 126.65, 126.54, 122.47, 122.25, 119.83, 115.39, 115.28, 63.80, 63.76, 50.91, 50.67, 48.68, 48.57, 45.42, 45.40, 44.96, 32.75, 28.86, 28.73. ESI MS: m/z = 730.1 [M+H]+ (100 %).
19-(4-(4-(2-Fluorophenyl)piperazin-1-yl)butyl)-1,16-diphenyl-19-azahexacyclo-[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (7)
Yield: 87 %, m.p. 205–207 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.83 (d, 2H, CHarom., J = 8.4 Hz), 8.28 (d, 2H, CHarom., J = 7.2 Hz), 7.74 (t, 2H, CHarom., J = 7.2 Hz), 7.58–7.52 (m, 4H, CHarom.), 7.42 (t, 2H, CHarom., J = 7.8 Hz), 7.24–7.14 (m, 4H, CHarom.), 7.10–6.95 (m, 6H, CHarom.), 4.68 (s, 2H, CH), 3.39–3.36 (m, 2H, CH2), 3.11–3.07 (m, 2H, CH2), 3.03–2.93 (m, 4H, CH2), 2.73–2.71 (m, 4H, CH2), 2.14–2.10 (m, 4H, CH2). 13C NMR (DMSO-d 6) δ (ppm): 197.20, 173.41, 173.35, 157.56, 147.54, 137.61, 134.41, 133.87, 133.79, 133.54, 133.49, 132.28, 132.17, 132.08, 132.02, 131.90, 131.76, 131.61, 131.55, 130.40, 130.17, 129.93, 129.82, 129.73, 129.70, 128.53, 128.34, 127.82, 126.69, 126.51, 122.48, 122.23, 119.88, 115.33, 115.27, 63.81, 63.74, 50.98, 50.63, 48.62, 48.54, 45.43, 45.41, 44.96, 32.72, 28.82, 28.79. ESI MS: m/z = 714.2 [M+H]+ (100 %).
19-(4-(4-(4-Acetylphenyl)piperazin-1-yl)butyl)-1,16-diphenyl-19-azahexacyclo-[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (8)
Yield: 77 %, m.p. 202–204 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.82 (d, 2H, CHarom., J = 8.1 Hz), 8.28 (d, 2H, CHarom., J = 7.8 Hz), 7.80–7.72 (m, 4H, CHarom.), 7.54 (t, 2H, CHarom., J = 7.2 Hz), 7.42 (t, 2H, CHarom., J = 7.5 Hz), 7.22 (t, 2H, CHarom., J = 7.8 Hz), 7.15 (d, 2H, CHarom., J = 7.8 Hz), 7.03 (d, 2H, CHarom., J = 8.1 Hz), 6.92 (d, 2H, CHarom., J = 9.3 Hz), 4.68 (s, 2H, CH), 3.52–3.44 (m, 4H, CH2), 3.16 (t, 4H, CH2, J = 4.2 Hz), 2.77 (t, 2H, CH2, J = 6.9 Hz), 2.44 (s, 3H, COCH3), 2.10–2.07 (m, 4H, CH2), 1.46 (t, 2H, CH2, J = 6.9 Hz). 13C NMR (DMSO-d 6) δ (ppm): 197.23, 186.59, 173.39, 173.37, 157.51, 147.59, 137.62, 134.57, 133.89, 133.85, 133.69, 133.57, 132.55, 132.34, 132.17, 132.11, 131.92, 131.85, 131.69, 131.57, 130.46, 130.38, 129.90, 129.83, 129.77, 129.72, 128.59, 128.30, 127.75, 126.61, 126.54, 122.47, 122.26, 119.82, 115.36, 115.34, 63.85, 63.82, 50.78, 50.68, 48.61, 48.59, 45.47, 45.44, 44.92, 32.77, 28.90, 28.83, 26.82. ESI MS: m/z = 738.6 [M+H]+ (100 %).
1,16-Diphenyl-19-(4-(4-(2-(trifluoromethyl)phenyl)piperazin-1-yl)butyl)-19-azahexa-cyclo[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (9)
Yield: 84 %, m.p. 211–212 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.78 (d, 2H, CHarom., J = 8.4 Hz), 8.30 (d, 2H, CHarom., J = 7.8 Hz), 7.74 (t, 2H, CHarom., J = 6.3 Hz), 7.69–7.60 (m, 3H, CHarom.), 7.54 (t, 3H, CHarom., J = 6.3 Hz), 7.48–7.40 (m, 4H, CHarom.), 7.18–7.14 (m, 2H, CHarom.), 4.48 (s, 2H, CH), 3.95–3.91 (m, 3H, CH2), 3.61–3.37 (m, 10H, CH2), 3.22–3.17 (m, 3H, CH2), 3.01–2.92 (m, 4H, CH2). 13C NMR (DMSO-d 6) δ (ppm): 197.19, 173.12, 173.05, 157.51, 147.74, 137.40, 134.36, 133.88, 133.77, 133.43, 133.37, 132.15, 132.10, 132.04, 132.01, 131.99, 131.78 (2C), 131.54, 130.48, 130.13, 129.92, 129.86, 129.71 (2C), 128.53, 128.37, 127.86, 126.66, 126.51, 123.92, 122.45, 122.18, 119.83, 115.34, 115.28, 63.80, 63.78, 61.17, 50.92, 50.68, 48.62, 48.59, 45.44, 45.41, 44.97, 32.76, 31.28, 28.87, 28.73. ESI MS: m/z = 792.2 [M+H]+ (100 %).
10-Diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (10)
The mixture of 2.06 g (0.006 mol) of 1,3-diphenylcyclopenta[a]indene-2,8-dione (“Indanocyclone”) was suspended in 75 ml of benzene and 0.65 g (0.006 mol) of maleimide was added. After refluxing time of 16 h the yellow residue was evaporated. Next it was purified by column chromatography (chloroform:methanol 9.5:0.5 vol). The combined fractions were condensed to dryness to give 1.50 g (73 %) of (10), m.p. 223–225 °C. 1H NMR (CDCl3) δ (ppm): 7.60 (d, 2H, CHarom., J = 2.7 Hz), 7.59–7.58 (m, 2H, CHarom.), 7.52 (d, 2H, CHarom., J = 2.1 Hz), 7.51–7.49 (m, 2H, CHarom.), 7.45 (d, 2H, CHarom., J = 2.1 Hz), 7.44–7.40 (m, 4H, CHarom.). 13C NMR (CDCl3) δ (ppm): 190.91, 165.89, 165.73, 149.69, 141.97, 139.37, 135.58, 135.52, 135.14, 134.81, 134.24, 131.59, 130.57, 130.54, 129.87, 129.34, 129.28 (2C), 129.09 (3C), 128.59 (2C), 127.91 (2C), 124.59, 124.54. ESI MS: m/z = 424.1 [M+Na]+ (100 %).
2-(4-Bromobutyl)-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (11)
A mixture of imide (10) (2.64 g, 0.006 mol), 1,4-dibromobutane (1.5 ml, 0.012 mol), anhydrous K2CO3 (2.51 g), and catalytic amount of KI were refluxed in acetonitrile for 14 h. Then the solvent was removed on a rotary evaporator and the dark yellow solid residue was purified by column chromatography (chloroform:methanol 9.5:0.5 vol). The combined fractions were condensed to dryness to give 2.44 g (92 %) of (11), m.p. 241–242 °C. 1H NMR (CDCl3) δ (ppm): 7.63–7.59 (m, 3H, CHarom.), 7.56–7.55 (m, 2H, CHarom.), 7.53–7.50 (m, 4H, CHarom.), 7.48–7.41 (m, 5H, CHarom.), 3.72 (q, 2H, CH2, J = 7.2 Hz), 3.54 (t, 2H, CH2, J = 6.9 Hz), 3.35 (t, 2H, CH2, J = 6.3 Hz), 1.26–1.21 (m, 2H, CH2). 13C NMR (CDCl3) δ (ppm): 190.21, 165.67, 165.49, 148.11, 141.34, 137.49, 135.09, 134.80, 134.26, 134.04, 133.87, 132.08, 130.52, 129.75, 129.37 (3C), 128.79 (3C), 128.51 (2C), 128.17, 127.14 (2C), 124.03, 123.48, 36.63, 34.50, 29.57, 26.48. ESI MS: m/z = 560.1 [M+Na]+ (100 %).
General method for the preparation of arylpiperazine derivatives of 2-(4-bromobutyl)-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (12–19)
A mixture of derivative (11) (0.3 g, 0.0005 mol) and the corresponding amine (0.001 mol), anhydrous K2CO3 (0.3 g), and catalytic amount of KI were refluxed in acetonitrile for 30 h. Then the mixture was filtered off and the solvent was evaporated. The yellow residue was purified by column chromatography (chloroform:methanol 9.5:0.5 vol) and/or crystallized from methanol. Obtained compounds were converted into their hydrochlorides. The solid product was dissolved in methanol saturated with gaseous HCl. The hydrochloride was precipitated by addition of diethyl ether. The crude product was crystallized from appropriate solvent.
4,10-Diphenyl-2-[4-(4-phenylpiperazin-1-yl)butyl]-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (12)
Yield: 87 %, m.p. 231–232 °C. 1H NMR (DMSO-d 6) δ (ppm): 7.61 (t, 3H, CHarom., J = 3.6 Hz), 7.56–7.44 (m, 8H, CHarom.), 7.40–7.31 (m, 2H, CHarom.), 7.28–7.23 (m, 2H, CHarom.), 6.98 (d, 2H, CHarom., J = 8.1 Hz), 6.86 (t, 1H, CHarom., J = 7.2 Hz), 6.23 (d, 1H, CHarom., J = 6.6 Hz), 3.76 (d, 2H, CH2, J = 11.4 Hz), 3.49–3.42 (m, 4H, CH2), 3.15–3.02 (m, 6H, CH2), 1.72–1.69 (m, 2H, CH2), 1.57–1.52 (m, 3H, CH2). 13C NMR (CDCl3) δ (ppm): 190.32, 165.58, 165.37, 149.52, 148.83, 141.58, 137.54, 135.13, 134.77, 134.39, 134.12, 133.94, 132.22, 130.47, 129.63 (2C), 129.41 (4C), 128.85 (2C), 128.49 (4C), 128.36 (2C), 127.24 (3C), 124.11, 123.53, 57.84, 57.65, 50.97, 50.86, 36.63, 34.50, 29.57, 26.48. ESI MS: m/z = 618.4 [M+H]+ (100 %).
4,10-Diphenyl-2-{4-[4-(pyridin-2-yl)piperazin-1-yl]butyl}-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (13)
Yield: 90 %, m.p. 219–220 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.14 (d, 1H, CHarom., J = 3.9 Hz), 7.82–7.74 (m, 1H, CHarom.), 7.61 (t, 3H, CHarom., J = 3.6 Hz), 7.56–7.48 (m, 8H, CHarom.), 7.40–7.31 (m, 2H, CHarom.), 7.19–7.02 (m, 1H, CHarom.), 6.84 (t, 1H, CHarom., J = 6.0 Hz), 6.23 (d, 1H, CHarom., J = 6.9 Hz), 4.37 (d, 2H, CH2, J = 15.0 Hz), 3.52–3.31 (m, 6H, CH2), 3.06–2.99 (m, 4H, CH2), 1.68–1.67 (m, 2H, CH2), 1.56–1.55 (m, 2H, CH2). 13C NMR (CDCl3) δ (ppm): 190.02, 165.63, 165.27, 153.84, 147.79, 141.44, 137.41, 135.58, 134.62, 134.29, 134.07, 133.68, 132.15, 130.32, 129.46 (2C), 129.39 (3C), 128.69 (2C), 128.38 (3C), 128.28, 128.20 (2C), 127.17 (3C), 124.46, 123.74, 52.35, 51.98, 48.79, 58.23, 36.96, 34.86, 27.62, 26.13. ESI MS: m/z = 619.4 [M+H]+ (100 %).
4,10-Diphenyl-2-[4-(4-{2-[2-(trifluoromethyl)phenyl]ethyl}piperazin-1-yl)butyl]-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (14)
Yield: 91 %, m.p. 232–233 °C. 1H NMR (DMSO-d 6) δ (ppm): 7.46–7.65 (m, 2H, CHarom.), 7.62–7.60 (m, 4H, CHarom.), 7.56–7.44 (m, 9H, CHarom.), 7.41–7.30 (m, 2H, CHarom.), 6.23 (d, 1H, CHarom., J = 6.6 Hz), 3.46–3.37 (m, 4H, CH2), 3.34–3.09 (m, 14H, CH2), 1.66–1.65 (m, 2H, CH2), 1.57–1.55 (m, 2H, CH2). 13C NMR (CDCl3) δ (ppm): 190.18, 166.94, 166.57, 149.97, 148.22, 142.53, 137.16, 135.86, 134.51, 134.72, 134.58, 134.02 (2C), 132.48, 131.30, 129.64 (2C), 129.58 (3C), 129.48, 128.91 (2C), 128.75 (3C), 128.59, 128.27 (2C), 127.85 (2C), 127.03, 124.59, 123.61, 123.00, 59.89, 55.12, 55.01, 50.79, 50.83, 36.74, 34.95, 32.73, 29.48, 26.17. ESI MS: m/z = 714.3 [M+H]+ (100 %).
2-{4-[4-(2-Fluorophenyl)piperazin-1-yl]butyl}-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (15)
Yield: 88 %, m.p. 245–247 °C. 1H NMR (DMSO-d 6) δ (ppm): 7.61 (t, 3H, CHarom., J = 3.9 Hz), 7.56–7.44 (m, 8H, CHarom.), 7.41–7.30 (m, 2H, CHarom.), 7.21–7.00 (m, 4H, CHarom.), 6.23 (d, 1H, CHarom., J = 7.8 Hz), 3.50–3.37 (m, 8H, CH2), 3.21–3.08 (m, 4H, CH2), 1.70–1.68 (m, 2H, CH2), 1.58–1.53 (m, 2H, CH2). 13C NMR (CDCl3) δ (ppm): 191.47, 166.12, 165.97, 149.48, 148.57, 141.72, 137.16, 135.69, 134.38, 134.21, 134.09, 133.92, 132.46, 130.85 (2C), 129.36 (2C), 129.29 (3C), 128.63 (2C), 128.52 (3C), 128.47 (2C), 127.69 (4C), 124.82, 123.96, 57.06, 56.93, 50.46, 50.27, 36.12, 34.98, 29.58, 26.02. ESI MS: m/z = 636.4 [M+H]+ (100 %).
2-{4-[4-(4-Fluorophenyl)piperazin-1-yl]butyl}-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (16)
Yield: 93 %, m.p. 241–242 °C. 1H NMR (DMSO-d 6) δ (ppm): 7.61 (t, 3H, CHarom., J = 3.9 Hz), 7.56–7.53 (m, 1H, CHarom.), 7.51–7.48 (m, 3H, CHarom.), 7.47–7.46 (m, 5H, CHarom.), 7.41–7.30 (m, 2H, CHarom.), 7.13–7.07 (m, 2H, CHarom.), 7.03–6.98 (m, 2H, CHarom.), 3.67 (d, 2H, CH2, J = 9.0 Hz), 3.47–3.42 (m, 4H, CH2), 3.06 (d, 6H, CH2, J = 8.4 Hz), 1.69–1.68 (m, 2H, CH2), 1.57–1.54 (m, 2H, CH2). 13C NMR (CDCl3) δ (ppm): 191.19, 166.58, 165.74, 149.53, 148.82, 141.13, 137.64, 135.97, 134.27, 134.09, 134.01, 133.84, 132.16, 130.76 (2C), 129.94 (3C), 129.59 (2C), 128.89 (3C), 128.72 (3C), 128.11 (2C), 127.75 (3C), 125.49, 123.52, 57.68, 57.51, 50.94, 50.00, 36.81, 34.86, 29.37, 26.97. ESI MS: m/z = 636.4 [M+H]+ (100 %).
2-{4-[4-(4-Chlorophenyl)piperazin-1-yl]butyl}-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (17)
Yield: 82 %, m.p. 248–249 °C. 1H NMR (DMSO-d 6) δ (ppm): 7.61 (t, 3H, CHarom., J = 3.6 Hz), 7.56–7.53 (m, 1H, CHarom.), 7.51–7.48 (m, 2H, CHarom.), 7.47–7.45 (m, 5H, CHarom.), 7.40–7.30 (m, 2H, CHarom.), 7.31–7.27 (m, 2H, CHarom.), 7.00 (d, 2H, CHarom., J = 9.0 Hz), 6.23 (d, 1H, CHarom., J = 7.5 Hz), 3.77 (d, 2H, CH2, J = 10.8 Hz), 3.49–3.72 (m, 4H, CH2), 3.07–3.01 (m, 6H, CH2), 1.68–1.66 (m, 2H, CH2), 1.57–1.52 (m, 2H, CH2). 13C NMR (CDCl3) δ (ppm): 190.64, 165.27, 165.11, 149.82, 148.56, 141.93, 137.14, 135.70, 134.31, 134.27, 134.03, 133.91, 132.27 (2C), 130.39 (2C), 129.79 (2C), 129.51 (3C), 128.88 (3C), 128.68 (3C), 128.02 (2C), 127.57 (2C), 124.69, 123.24, 57.49, 57.33, 50.17, 50.06, 36.94, 34.42, 29.96, 26.76. ESI MS: m/z = 652.4 [M+H]+ (100 %).
2-{4-[4-(2-Chlorophenyl)piperazin-1-yl]butyl}-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (18)
Yield: 75 %, m.p. 235–237 °C. 1H NMR (DMSO-d 6) δ (ppm): 7.60 (t, 3H, CHarom., J = 3.6 Hz), 7.56–7.55 (m, 1H, CHarom.), 7.53–7.48 (m, 2H, CHarom.), 7.47–7.44 (m, 6H, CHarom.), 7.40–7.31 (m, 3H, CHarom.), 7.20–7.08 (m, 2H, CHarom.), 6.23 (d, 1H, CHarom., J = 7.8 Hz), 3.51–3.28 (m, 6H, CH2), 3.19–3.07 (m, 6H, CH2), 1.70–1.68 (m, 2H, CH2), 1.58–1.53 (m, 6H, CH2). 13C NMR (CDCl3) δ (ppm): 190.30, 165.71, 165.49, 149.83, 148.79, 141.26, 137.44, 135.86, 134.92, 134.77, 134.51, 133.34 (2C), 132.58 (2C), 130.93 (2C), 129.81 (2C), 129.79 (2C), 128.73 (3C), 128.52 (3C), 128.39 (2C), 127.04 (2C), 124.82, 123.17, 58.14, 58.07, 52.58, 52.47, 35.97, 34.06, 29.74, 26.11. ESI MS: m/z = 652.4 [M+H]+ (100 %).
Synthesis of 2-{4-[4-(2-metoxyphenyl)piperazin-1-yl]butyl}-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (19)
Yield: 79 %, m.p. 245–246 °C. 1H NMR (DMSO-d 6) δ (ppm): 7.61 (t, 3H, CHarom., J = 3.6 Hz), 7.56–7.44 (m, 8H, CHarom.), 7.41–7.31 (m, 2H, CHarom.), 7.05–6.87 (m, 4H, CHarom.), 6.23 (d, 1H, CHarom., J = 6.9 Hz), 3.79 (s, 3H, OCH3), 3.47–3.44 (m, 6H, CH2), 3.07–2.97 (m, 6H, CH2), 1.69–1.67 (m, 2H, CH2), 1.59–1.52 (m, 2H, CH2). 13C NMR (CDCl3) δ (ppm): 192.35, 165.07, 164.79, 149.81, 148.96, 141.13, 137.77, 135.42, 134.37, 134.26, 134.08, 133.11 (2C), 132.66 (2C), 130.72 (3C), 129.86, 129.72 (2C), 128.91 (3C), 128.54 (2C), 128.21 (3C), 127.75 (2C), 124.11, 123.59, 62.00, 58.84, 58.71, 52.97, 52.84, 35.06, 34.26, 29.59, 26.91. ESI MS: m/z = 648.3 [M+H]+ (100 %).
3-{4-[4-(2-Metoxyphenyl)piperazin-1-yl]butyl}3-azatricyclo[7.3.1.05,13]trideca-(12),5,7,9(13),10-pentaene-2,4-dione (20) was obtained according to method presented previously (Hackling et al., 2003)
Yield: 63 %, m.p. 279–282 °C. 1H NMR (DMSO-d 6) δ (ppm): 8.59–8.48 (d, 2H, CHarom., J = 8.1 Hz), 8.11 (d, 2H, CHarom., J = 7.8 Hz), 7.64 (t, 2H, CHarom., J = 7.6 Hz), 7.08–6.76 (m, 4H, CHarom.) 4.56–4.17 (m, 2H, CH2), 3.87 (s, 3H, OCH3), 3,41–2.98 (m, 5H, CH2), 2.93–2.32 (m, 5H, CH2), 2.04–1.42 (m, 4H, CH2). 13C NMR (CDCl3) δ (ppm): 165.72, 159.08, 158.97, 140.62, 134.22, 134.17, 134.09, 133.74, 132.25, 130.14, 129.64, 129.53, 128.47, 128.38, 128.09, 127.48, 124.02, 123.61, 61.13, 60.95, 57.53, 51.27, 51.13, 41.37, 41.29, 26.96, 26.87. ESI MS: m/z = 344.6 [M+H]+ (100 %).
Biological assays
Cell-based assays
Cell-based assays were performed at Dipartimento di Scienze e Tecnologie Biomediche, Università di Cagliari, Monserrato, Italy.
Test compounds
Compounds were dissolved in DMSO at 100 mM and then diluted in culture medium.
Cells and viruses
Cell line and viruses were purchased from the American Type Culture Collection (ATCC). The absence of mycoplasma contamination was checked periodically by the Hoechst staining method. Cell line supporting the multiplication of human immunodeficiency virus type-1 (HIV-1) was the CD4+ human T-cells containing an integrated HTLV-1 genome (MT-4).
Cytotoxicity assays
Cytotoxicity assays were run in parallel with antiviral assays.
Exponentially growing MT-4 cells were seeded at an initial density of 1 × 105 cells/ml in 96-well plates in RPMI-1640 medium, supplemented with 10 % fetal bovine serum (FBS), 100 units/ml penicillin G, and 100 μg/ml streptomycin. Cell cultures were then incubated at 37 °C in a humidified 5 % CO2 atmosphere in the absence or presence of serial dilutions of test compounds. Cell viability was determined after 96 h at 37 °C by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) method (Pauwels et al., 1988).
Antiviral assays
Compound’s activity against HIV-1 was based on inhibition of virus-induced cytopathogenicity in MT-4 cell acutely infected with a multiplicity of infection (m.o.i.) of 0.01. In brief, 50 μl of RPMI containing 1 × 104 MT-4 cells were added to each well of flat-bottom microtitre trays, containing 50 μl of RPMI with or without serial dilutions of test compounds. Then, 20 μl of a HIV-1 suspension containing 100 CCID50 was added. After a 4-day incubation at 37 °C, cell viability was determined by the MTT method (Pauwels et al., 1988).
In vitro ligand binding assays
Ligand studies with native 5-HT1A receptor were conducted according to the methods previously described (Lewgowd et al., 2011).
X-ray structure determination
Suitable crystals were mounted for measurements. The X-ray measurements were performed at 100(2) K on a KUMA CCD k-axis diffractometer with graphite-monochromated Mo Kα radiation (0.71073 Å). The crystals were positioned at 62.25 mm from the KM4CCD camera. The data were corrected for Lorentz and polarization effects, additionally absorption corrections were applied. Data reduction and analysis were carried out with the Kuma Diffraction (Wrocław, Poland) programmes (Oxford Diffraction CrysAlis CCD and CrysAlis RED, 2001). The structures were solved by direct methods (Sheldrick, 1990) and refined by using SHELXL (Sheldrick, 1997) The refinement was based on F 2 for all reflections except for those with very negative F 2. The weighted R factor, wR, and all goodness-of-fit S values are based on F 2. The non-hydrogen atoms were refined anisotropically. The hydrogen atoms were located from a difference map and were refined isotropically. The atomic scattering factors were taken from the International Tables (Wilson, 1992). Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 913714-913719. Copy of the data can be obtained on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (email: deposit@ccdc.cam.ac.uk).
X-ray crystal data for 2
C37H28BrNO3, monoclinic space group P21/c: a = 15.7066(8), b = 7.9750(4), c = 23.0807(12) Å, β = 100.366(4); V = 2843.9(3) Å3, Z = 4, D calcd = 1.435 g/cm3; μ = 1.485 mm−1; F(000) = 1264. A total of 21,137 reflections were integrated in the θ-range of 2.71°–25.0° of which 5,007 were unique, leaving an overall R-merge of 0.041. For solution and refinement, 5,007 were considered as unique after merging for Fourier. The final agreement factors were R1 = 0.028 for 3,431 reflections with F > 4σ(F); R1 = 0.0501 and wR2 = 0.0553 for all the 5,007 data; GOF = 0.864. The residual electron density in the final difference Fourier does not show any feature above 0.33 e Å−3 and below −0.32 e Å−3.
X-ray crystal data for 6
C47H40ClN3O3, monoclinic space group P21/n: a = 11.8478(9), b = 23.8155(18), c = 13.0659(10) Å, β = 101.732(6); V = 3609.7(5) Å3, Z = 4, D calcd = 1.344 g/cm3; μ = 0.155 mm−1; F(000) = 1536. A total of 27,540 reflections were integrated in the θ-range of 2.72°–25.0° of which 6,356 were unique, leaving an overall R-merge of 0.0653. For solution and refinement, 6,348 were considered as unique after merging for Fourier. The final agreement factors were R1 = 0.0339 for 2,916 reflections with F > 4σ(F); R1 = 0.0935 and wR2 = 0.1195 for all the 6348 data; GOF = 0.854. The residual electron density in the final difference Fourier does not show any feature above 0.22 e Å−3 and below −0.22 e Å−3.
X-ray crystal data for 7
C47H40FN3O3, monoclinic space group P21/n: a = 11.8103(4), b = 23.4267(5), c = 13.2359(3) Å, β = 96.196(2); V = 3640.67(17) Å3, Z = 4, D calcd = 1.302 g/cm3; μ = 0.085 mm−1; F(000) = 1504. A total of 27,438 reflections were integrated in the θ-range of 2.8°–25.0° of which 6,394 were unique, leaving an overall R-merge of 0.0104. For solution and refinement, 6,394 were considered as unique after merging for Fourier. The final agreement factors were R1 = 0.0323 for 5,658 reflections with F > 4σ(F); R1 = 0.0365 and wR2 = 0.1276 for all the 6,394 data; GOF = 1.144. The residual electron density in the final difference Fourier does not show any feature above 0.24 e Å−3 and below −0.2 e Å−3.
X-ray crystal data for 11
C31H22BrNO3, monoclinic space group P21: a = 9.3851(7), b = 23.3058(14), c = 11.4605(7) Å, β = 106.711(7); V = 2400.9(3) Å3, Z = 4, D calcd = 1.484 g/cm3; μ = 1.747 mm−1; F(000) = 1,096. A total of 9,877 reflections were integrated in the θ-range of 2.86°–26.0° of which 6,914 were unique, leaving an overall R-merge of 0.0318. For solution and refinement, 4,835 were considered as unique after merging for Fourier. The final agreement factors were R1 = 0.0633 for 4,665 reflections with F > 4σ(F); R1 = 0.1047 and wR2 = 0.1518 for all the 6,914 data; GOF = 1.049. The residual electron density in the final difference Fourier does not show any feature above 1.05 e Å−3 and below −0.96 e Å−3.
X-ray crystal data for 19
C41H36Cl2N3O3, triclinic space group P-1: a = 11.4607(3), b = 12.0127(3), c = 13.7081(4) Å, α = 97.455(2), β = 103.874(2), γ = 105.357(2); V = 1728.71(8) Å3, Z = 2, D calcd = 1.337 g/cm3; μ = 0.234 mm−1; F(000) = 728. A total of 19,541 reflections were integrated in the θ-range of 3.01°–25.0° of which 6,084 were unique, leaving an overall R-merge of 0.0173. For solution and refinement, 6,084 were considered as unique after merging for Fourier. The final agreement factors were R1 = 0.0351 for 4,789 reflections with F > 4σ(F); R1 = 0.0471 and wR2 = 0.0956 for all the 6,084 data; GOF = 1.077. The residual electron density in the final difference Fourier does not show any feature above 0.29 e Å−3 and below −0.25 e Å−3.
X-ray crystal data for 20
C27H30ClN3O3, triclinic space group P-1: a = 7.66540(10), b = 10.3318(2), c = 16.0440(3) Å, α = 96.0230(10), β = 93.910(2), γ = 106.740(2); V = 1203.60(4) Å3, Z = 2, D calcd = 1.324 g/cm3; μ = 0.193 mm−1; F(000) = 508. A total of 13,968 reflections were integrated in the θ-range of 2.94°–25.0° of which 4,235 were unique, leaving an overall R-merge of 0.0149. For solution and refinement, 4,235 were considered as unique after merging for Fourier. The final agreement factors were R1 = 0.0267 for 3,532 reflections with F > 4σ(F); R1 = 0.0327 and wR2 = 0.0758 for all the 4,235 data; GOF = 1.068. The residual electron density in the final difference Fourier does not show any feature above 0.27 e Å−3 and below −0.21 e Å−3.
Results and discussion
Chemistry
Synthesis of N-butylarylpiperazinyl derivatives
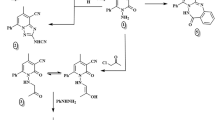
Two synthetic lines of N-substituted arylpiperazine derivatives were prepared. In the first path (Scheme 1), commercially available 1,3-diphenyl-2H-cyclopenta[l]phenanthren-2-one (“Phencyclone”) and maleimide were condensed in Diels–Alder reaction, and toluene was used as a solvent. After addition of 1,4-dibromobutane, 1,16-diphenyl-19-azahexacyclo[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione was obtained (1). Finally, synthesized 19-(4-bromobutyl)-1,16-diphenyl-19-azahexacyclo-[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (2) was used to obtain seven new complex arylpiperazines (3–9).

Synthesis of butylarylpiperazinyl derivatives of 1,16-diphenyl-19-azahexacyclo[14.5.1.02,15.03,8.09,14.017,21]docosa-2,3,5,7,8,9,11,13,14-nonaene-18,20,22-trione (1)
In the second synthetic path (Scheme 2), “Indanocyclone” and maleimide were refluxed to give 4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (10). This step of synthesis shows different approaches (decarbonylation) of the condensation reaction between dienes and dienophiles.

1,3-Diphenylcyclopenta[a]indene-2,8-dione as starting material for new synthetic route of complex arylpiperazines
The 2-(4-bromobutyl)-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (11) was obtained by condensation of 1,4-dibromobutane with above-mentioned complex imide in acetonitrile used as a solvent. The final step was to synthesize arylpiperazine derivatives by refluxing corresponding piperazines with 2-(4-bromobutyl)-4,10-diphenyl-1H,2H,3H,5H-indeno[1,2-f]isoindole-1,3,5-trione (11). Crude products (12–19) were purified and their hydrochlorides were made.
In addition, the synthesis of 3-{4-[4-(2-metoxyphenyl)piperazin-1-yl]butyl}3-azatricyclo-[7.3.1.05,13]trideca(12),5,7,9(13),10-pentaene-2,4-dione (20) was carried out. The compound was the subject of previous biological (5-HTR affinity) investigations (Scheme 3), however, the X-ray crystal analysis of the compound has not been published yet.

Synthesis of 3-{4-[4-(2-metoxyphenyl)piperazin-1-yl]butyl}3-azatricyclo[7.3.1.05,13]trideca-(12),5,7,9(13),10-pentaene-2,4-dione (20)
All obtained compounds were purified by flash chromatography. Elemental analysis, mass spectrometry, 1H NMR and 13C NMR spectra confirmed the identity of the products. For compounds 2 and 11, also for hydrochlorides of 6, 7, 19, and 20 X-ray analyses were done.
Biology
Cytotoxicity and anti HIV-1 activity
Title compounds were tested in cell-based assay against the human immunodeficiency virus type-1 (HIV-1), using Efavirenz as reference inhibitor. The cytotoxicity was evaluated in parallel with the antiviral activity.
None of tested compounds showed selective antiviral activity against HIV-1. However compounds 10 and 14 turned out cytotoxic for exponentially growing MT4 cells in the low micromolar range (CC50 = 9 μM) (Table 1).
X-ray structural analyses
The crystal structures have been determined for three “phencyclone” derivatives 2, 6, and 7. Their main skeleton resembles buspirone, but have more bulky maleimide fragment and in the case of 2 there is no piperazine moiety (n-butyl chain is terminated by bromine atom). In structures 6 and 7, the aromatic fragment (p-chlorophenyl and o-fluorophenyl, respectively) is different from 2-pirymidinyl substituent in buspirone.
In all of these structures phenanthrene moiety forms a kind of “roof” over n-butyl chain, and phenyl rings are situated like “wings” directed outside (Fig. 2). In structures 6 and 7, the piperazine moiety adopts chair conformation. All compounds crystallize in monoclinic system without solvent with one molecule in an asymmetric unit. Unit cell contains 4 molecules related by inversion center (Fig. 3).

Crystal structures of 2, 6, and 7. Thermal ellipsoids drawn at 50 % probability level

Crystal packing of 2, 6, and 7
The crystal structure of 2 is stabilized by two kinds of short interactions between C–H···O and C–H···Br (Fig. 4). In 6 there are three types of C–H···O contacts. The oxygen atom from maleimide moiety contacts with piperazine and phenanthrene fragments. Second one interacts with phenyl ring (Fig. 5). The structure of 7 shows similar C–H···O interactions and there is an additional short C–H···F contact (Fig. 6).

Short intermolecular contacts in crystal structure of 2

Short intermolecular contacts in crystal structure of 6

Short intermolecular contacts in crystal structure of 7
Two crystal structures based on “Indanocyclone” 11 and 19 are disordered. Compound 11 crystallizes without solvent in monoclinic P21 space group with two molecules in an asymmetric unit. The structure is a racemic twin in which one molecule is disordered. The disorder occurs in the n-butyl chain together with bromine atom and in the first phenyl ring of Indanocyclone. Two side phenyl rings are almost coplanar, the angle between mean best planes is 3.5°. There are three types of C–H···O interactions between maleimide oxygens and the n-butyl chain, as well as the side phenyl ring, and between oxygen from Indanocyclone moiety and the side phenyl ring (Fig. 7).

Crystal packing and short intermolecular contacts in crystal structure of 11
Compound 19 crystallizes as a hydrochloride with one molecule of water in triclinic P-1 space group with one molecule in an asymmetric unit. Disorder occurs in first Indanocyclone phenyl ring and gives rise to π···π stacking between disordered benzene and maleimide rings. Two side phenyl rings are tilted with respect to each other by 24.8° (Fig. 8). The n-butyl chain adopts cis conformation with dihedral angle N1-C28-C29-C30 equal to 55.6.

Crystal packing of 19. Disordered phenyl ring showing π···π stacking with maleimide ring
The structure is stabilized by a set of N+H···Cl− bonds between piperazine and chloride anions. There are two types of interactions between oxygens from maleimide moiety and C–H from butyl chain and Indanocyclone phenyl ring. Water molecule forms C–H···O bonds with piperazine and Indanocyclone phenyl ring. There are also O–H···Cl− interactions (Fig. 9).

Short intermolecular contacts in crystal structure of 19
Compound 20, an analog of NAN-190, crystallizes in triclinic P-1 space group as a hydrochloride with one molecule in an asymmetric unit. The imide moiety is almost planar. The piperazine ring adopts chair conformation (Fig. 10). The crystal structure forms layers along a axis comprising of alternating molecules (Fig. 11). The structure is stabilized by N+H···Cl− hydrogen bonds. In addition there are short contacts between chloride anion and C–H from the methoxy group, the butyl chain and the piperazine moiety. There are also interactions between oxygens from the imide fragment with C–H from piperazine and the methoxyphenyl ring (Fig. 12).

Crystal structure of 20. Thermal ellipsoids drawn at 50 % probability level

Crystal packing of 20. View along a axis

Short intermolecular contacts in crystal structure of 20
Conclusions
Compounds 6, 7, 19, and 20 fit well to the three-point pharmacophore model for 5-HT1A receptor ligands (Chilmonczyk et al., 1997). Apart from an aromatic ring and the basic nitrogen of piperazine, localized in the distance of 5.2–5.7 Å from a centroid, authors have found the third point essential for a ligand–receptor interaction—the carbonyl oxygen, expected in the distance of 7.07 Å from the center of an aromatic ring and 4.3 Å from N4 piperazine atom. Intramolecular distances measured for a set of 5-HT1A receptor ligands by Chilmonczyk et al. were in the range of 7.93–12.37 Å (Centroid···O(1)), 3.95–7.16 Å (N(1)···O(1)), and 5.15–5.64 Å (Centroid···N(1)).
The values calculated for new arylpiperazine derivatives (6, 7, 19, and 20) are in agreement with the presented three-point pharmacophore model (Table 2, Fig. 13). The distance between the center of the phenyl group and the imide oxygen (O1) is in the range of 8,13–11,89 Å. The measured distance of the protonated nitrogen (N1) and O1 atom is in the range of 4.06–6.66 Å. The value of centroid –N1 length is in a narrow range between 5.67 and 5.71 Å. Presented results suggest that compounds 6, 7, 19, and 20 could serve as potential 5-HT1A receptor ligands. They also prove that similar molecular values can be estimated for the derivative 4. Although it is an exception from “the rule of five,” because of its high molecular weight, volume and logP, and low solubility logS (Table 3), the compound 4 possess moderate activity to the 5-HT1A receptor.

Molecular geometric parameters (in Å) observed in solid state for the derivative 20
Structural data obtained for a set of long-chain arylpiperazine derivatives can serve for further investigations concerning ligands activity to metabotropic 5-HT receptors.
References
Ananda Kumar CS, Benaka Prasad SB, Vinaya K, Chandrappa S, Thimmegowda NR, Kumar YC, Swarup S, Rangappa KS (2009) Synthesis and in vitro antiproliferative activity of novel 1-benzhydrylpiperazine derivatives against human cancer cell lines. Eur J Med Chem 44:1223–1229
Bojarski AJ (2006) Pharmacophore models for metabotropic 5-HT receptor ligands. Curr Top Med Chem 6:2005–2026
Bronowska A, Leś A, Chilmonczyk Z, Filipek S, Edvardsen O, Ostensen R, Sylte I (2001) Molecular dynamics of buspirone analogues interacting with the 5-HT1A and 5-HT2A serotonin receptors. Bioorg Med Chem 9:881–895
Chilmonczyk Z, Szelejewska-Wozniakowska A, Cybulski J, Cybulski M, Koziol AE, Gdaniec M (1997) Conformational flexibility of serotonin1A receptor ligands from crystallographic data. Updated model of the receptor pharmacophore. Arch Pharm (Weinheim) 330:146–160
Czopek A, Byrtus H, Kołaczkowski M, Pawłowski M, Dybała M, Nowak G, Tatarczyńska E, Wesołowska A, Chojnacka-Wójcik E (2010) Synthesis and pharmacological evaluation of new 5-(cyclo)alkyl-5-phenyl- and 5-spiroimidazolidine-2,4-dione derivatives. Novel 5-HT1A receptor agonist with potential antidepressant and anxiolytic activity. Eur J Med Chem 45:1295–1303
Filosa R, Peduto A, de Caprariis P, Saturnino C, Festa M, Petrella A, Pau A, Pinna GA, La Colla P, Busonera B, Loddo R (2007) Synthesis and antiproliferative properties of N3/8-disubstituted 3,8-diazabicyclo[3.2.1]octane analogues of 3,8-bis[2-(3,4,5-trimethoxyphenyl)pyridin-4-yl]methyl-piperazine. Eur J Med Chem 42:293–306
González-Gómez JC, Santana L, Uriarte E, Brea J, Villazón M, Loza MI, De Luca M, Rivas ME, Montenegro GY, Fontenla JA (2003) New arylpiperazine derivatives with high affinity for alpha1A, D2 and 5-HT2A receptors. Bioorg Med Chem Lett 13:175–178
Hackling A, Ghosh R, Perachon S, Mann A, Höltje HD, Wermuth CG, Schwartz JC, Sippl W, Sokoloff P, Stark H (2003) N-(omega-(4-(2-methoxyphenyl)piperazin-1-yl)alkyl)carboxamides as dopamine D2 and D3 receptor ligands. J Med Chem 46:3883–3899
Kerns EH, Di L (2008) Drug-like properties: concepts structure design and methods: from ADME to toxicity optimization. Academic Press, Amsterdam
Kim MK, Lee HS, Kim S, Cho SY, Roth BL, Chong Y, Choo H (2012) 4-Aminoethylpiperazinyl aryl ketones with 5-HT1A/5-HT7 selectivity. Bioorg Med Chem 20:1139–1148
Klabunde T, Evers A (2005) GPCR antitarget modeling: pharmacophore models for biogenic amine binding GPCRs to avoid GPCR-mediated side effects. ChemBioChem 6:876–889
Leopoldo M (2004) Serotonin(7) receptors (5-HT(7)Rs) and their ligands. Curr Med Chem 11:629–661
Lepailleur A, Bureau R, Paillet-Loilier M, Fabis F, Saettel N, Lemaître S, Dauphin F, Lesnard A, Lancelot JC, Rault S (2005) Molecular modeling studies focused on 5-HT7 versus 5-HT1A selectivity. Discovery of novel phenylpyrrole derivatives with high affinity for 5-HT7 receptors. J Chem Inf Model 45:1075–1081
Lewgowd W, Bojarski AJ, Szczesio M, Olczak A, Glowka ML, Mordalski S, Stanczak A (2011) Synthesis and structural investigation of some pyrimido[5,4-c]quinolin-4(3H)-one derivatives with a long-chain arylpiperazine moiety as potent 5-HT(1A/2A) and 5-HT(7) receptor ligands. Eur J Med Chem 46:3348–3361
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25
López-Rodríguez ML, Porras E, Morcillo MJ, Benhamú B, Soto LJ, Lavandera JL, Ramos JA, Olivella M, Campillo M, Pardo L (2003) Optimization of the pharmacophore model for 5-HT7R antagonism. Design and synthesis of new naphtholactam and naphthosultam derivatives. J Med Chem 46:5638–5650
Nowak M, Kołaczkowski M, Pawłowski M, Bojarski AJ (2006) Homology modeling of the serotonin 5-HT1A receptor using automated docking of bioactive compounds with defined geometry. J Med Chem 49:205–214
Oprea TI (2002) Virtual screening in lead discovery: a viewpoint. Molecules 7:51–62
Oxford Diffraction Poland (2001) Oxford Diffraction CrysAlis CCD and CrysAlis RED. Oxford Diffraction Poland, Wrocław
Pardo L, Deupi X, Dölker N, López-Rodríguez ML, Campillo M (2007) The role of internal water molecules in the structure and function of the rhodopsin family of G protein-coupled receptors. ChemBioChem 8:19–24
Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E (1988) Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods 20:309–321
Prandi A, Franchini S, Manasieva LI, Fossa P, Cichero E, Marucci G, Buccioni M, Cilia A, Pirona L, Brasili L (2012) Synthesis biological evaluation and docking studies of tetrahydrofuran- cyclopentanone- and cyclopentanol-based ligands acting at adrenergic α1- and serotonine 5-HT1A receptors. J Med Chem 55:23–36
Roth BL, Choudhary MS, Khan N, Uluer AZ (1997) High-affinity agonist binding is not sufficient for agonist efficacy at 5-hydroxytryptamine2A receptors: evidence in favor of a modified ternary complex model. J Pharmacol Exp Ther 280:576–583
Sheldrick GM (1990) Phase annealing in SHELX-90: direct methods for larger structures. Acta Cryst A 46:467–473
Sheldrick GM (1997) SHELXL97 program for the refinement of crystal structures. University of Göttingen, Göttingen
Siracusa MA, Salerno L, Modica MN, Pittalà V, Romeo G, Amato ME, Nowak M, Bojarski AJ, Mereghetti I, Cagnotto A, Mennini T (2008) Synthesis of new arylpiperazinylalkylthiobenzimidazole, benzothiazole, or benzoxazole derivatives as potent and selective 5-HT1A serotonin receptor ligands. J Med Chem 51:4529–4538
Sylte I, Bronowska A, Dahl SG (2001) Ligand induced conformational states of the 5-HT(1A) receptor. Eur J Pharmacol 416:33–41
Varin T, Gutiérrez-de-Terán H, Castro M, Brea J, Fabis F, Dauphin F, Aqvist J, Lepailleur A, Perez P, Burgueño J, Vela JM, Loza MI, Rodrigo J (2010) Phe369(738) at human 5-HT(7) receptors confers interspecies selectivity to antagonists and partial agonists. Br J Pharmacol 159:1069–1081
Vermeulen ES, Schmidt AW, Sprouse JS, Wikström HV, Grol CJ (2003) Characterization of the 5-HT(7) receptor. Determination of the pharmacophore for 5-HT(7) receptor agonism and CoMFA-based modeling of the agonist binding site. J Med Chem 46:5365–5374
Wilson AJC (1992) International tables for crystallography, vol C. Kluwer Academic Publishers, Dordrecht, pp 583–584
Yang L, Xu X, Huang Y, Zhang B, Zeng C, He H, Wang C, Hu L (2010) Synthesis of polyhydroxylated aromatics having amidation of piperazine nitrogen as HIV-1 integrase inhibitor. Bioorg Med Chem Lett 20:5469–5471
Acknowledgments
Authors are grateful to Professor Paolo La Colla (Universita di Cagliari, Monserrato, Italy) for performing cytotoxicity and HIV-1 activity screenings, and Professor Andrzej Bojarski (Institute of Pharmacology, Polish Academy of Science, Kraków, Poland) for 5-HT1A affinity investigation.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Szulczyk, D., Bielenica, A., Dobrowolski, M.A. et al. Synthesis and structure evaluation of new complex butylarylpiperazin-1-yl derivatives. Med Chem Res 23, 1519–1536 (2014). https://doi.org/10.1007/s00044-013-0740-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-013-0740-z