
Abstract
Series of 1-[2-thiazol-4-yl-(2-aminoethyl)]- and 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazine derivatives have been prepared and in vitro tested as H3-receptor antagonists (the electrically evoked contraction of the guinea-pig jejunum). It appeared that by comparison of homologous pairs, the 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazines (3a,b and 4a–d) have much higher potency than their analogous 1-[2-thiazol-4-yl-(2-aminoethyl)]-4-n-propylpiperazines (2a–k). Based on the obtained results, we observed the 5-position of 2-methyl-2-R-aminoethyl substituents in the thiazole ring is favourable for histamine H3 receptor antagonist activity, whereas its presence in position 4 leads, almost in each case, to strong decrease of activity.
Similar content being viewed by others
Introduction
Histamine plays a variety of physiological roles in the central nervous system (CNS) and peripheral tissues through the four known G protein-coupled receptors, H1, H2, H3 and H4 (Hough, 2001). H1 and H2 receptor antagonists are well-known therapeutic agents and are in use for the treatment of allergic disease (Leurs et al., 2002) and peptic ulcer (Brimblecombe et al., 1978), respectively. The newly discovered H4 receptor seems to have a role in regulating inflammatory responses (Thurmond et al., 2004). The histamine H3 receptor, which was discovered in 1983 by Arrang and co-workers (Arrang et al., 1983), mainly located in the CNS, is a presynaptic autoreceptor that does not only modulate the production and the release of histamine from histaminergic neurons (Arrang et al., 1987) but also regulates the release of other neurotransmitters like acetylocholine (Clapham and Kilpatrick, 1992; Yokatoni et al., 2000), dopamine (Schlicker et al., 1993), norepinephrine (Schlicker et al., 1990), serotonin (Schlicker et al., 1988) and glutamate (Brown and Reymann, 1996) in both the CNS and peripheral nervous system. Enhancement of neurotransmitter release by histamine H3 receptor antagonist shows a clinical approach to the treatment of several CNS disorders (Esbenshade et al., 2006; Cemkov et al., 2009), including attention deficit hyperactivity disorder (Quades, 1987), sleep disorders (Monti, 1993), epilepsy (Vahora et al., 2001) and schizophrenia (Velligan and Miller, 1999). Pharmacological data also suggest a potential role for H3 antagonists in the control of feeding, appetite, and support the role of H3 receptor in obesity (Hancock, 2003; Hancock et al., 2004).
Early generation of H3 receptor ligands were based on structures containing the imidazole moiety, many of which have found utility as pharmacological tools (Stark et al., 1996; Van der Goot and Timmerman, 2000). However, antagonist carrying on the imidazole heterocycle is the potential issue for drug–drug interactions through inhibition of hepatic cytochrome P450 enzymes and poor CNS penetration (Lin and Lu, 1998; Zhang et al., 2005). For these reasons, and after the successful cloning of the human histamine H3 receptor by Lovenberg (Lovenberg et al., 1999), efforts have been directed towards the discovery of H3 antagonists without an imidazole moiety as these compounds may offer improvements in binding affinity, CNS penetration, and reduced potential for cytochrome P450 enzymes inhibition (Cowart et al., 2004). A number of non-imidazole antagonists have since been reported (Ganellin et al., 1998; Celanire et al., 2005). Representative examples of non-imidazole H3 antagonists included among others were JNJ-5207852 (hH3RKi = 0.6 nM) (Apodaca et al., 2003), UCL 2190 (rH3RKi = 4 nM) (Meier et al., 2001) and ABT-239 (hH3RKi = 0.45 nM) (Cowart et al., 2002) (Chart 1).

Representative non-imidazole H3-histamine receptor antagonists and the target molecules of this study
Previously, our laboratory has described several non-imidazole piperazine-based histamine H3 antagonists, consisting of 1-(2-thiazolobenzo)-, 1-(2-thiazolopyridine)- and 1-[2-thiazol-5-yl-(2-aminoethyl)] moieties with moderate to pronounced affinity for the receptor (Walczyński et al., 1999, 2005; Frymarkiewicz and Walczynski, 2009). The SAR of 1-[(2-thiazolobenzo)-4-n-propyl]piperazines and 1-[(2-thiazolopyridine)-4-n-propyl]piperazines series, showed no significant difference in H3 activities (Walczyński et al., 1999, 2005). These results prompted us to replace the benzo ring by 2-methyl-2-alkylaminoethyl amide, 2-methyl-2-alkylaminoethyl and 2-methyl-2-phenylalkylaminoethyl chains at position 5 of 1-(2-thiazol-5-yl)-4-n-propylpiperazine moiety. The highest affinity for these series has been seen in the compound with the N-methyl-N-phenylpropylamino substituent 1 (Chart 1; pA2 = 8.27; electric field stimulation assay on guinea-pig jejunum) and with slightly lower potencies for compounds carrying on N-methyl-N-benzylamino and N,N-dimethylamino substituents with pA2 = 7.75 and 7.78, respectively (Frymarkiewicz and Walczynski, 2009).
In continuation of our earlier work, we studied the influence, on H3-receptor antagonistic activity, of the introduction of 2-CH3-2-R-aminoethyl-substitution at position 4 of the thiazole ring. Therefore, the series of 1-[2-thiazol-4-yl-(2-aminoethyl)]-4-n-propylpiperazines 2a–k (Chart 1), bearing the substituents showing the highest affinity in previously described 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazines (Frymarkiewicz and Walczynski, 2009), was prepared and pharmacologically evaluated (electric field stimulation assay on guinea-pig jejunum). In addition, with the aim of the complement 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazines series, 1-[2-thiazol-5-yl-(2-methyl-2-phenylethyl)]- 3a, 1-[2-thiazol-5-yl-(2-methyl-2-phenylbutylaminoethyl)]-4-n-propylpiperazine 3b and 1-[2-thiazol-5-yl-(2-methyl-2-phenylcarbonylaminoethyl)]-4-n-propylpiperazine amides 4a–d (Chart 1) were synthesized.
In this study, we report on synthesis and preliminary pharmacological investigation of new 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazine derivatives 2 and 1-[2-thiazol-5-yl-(2-methyl-2-phenylethyl-, 1-[2-thiazol-5-yl-(2-methyl-2-phenylbutylaminoethyl)]-4-n-propylpiperazines 3 and 1-[2-thiazol-5-yl-(2-methyl-2-phenylcarbonylaminoethyl)]-4-n-propylpiperazine amides 4.
Chemistry
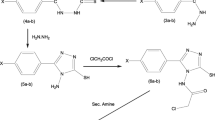
The general synthetic procedure used in this study is illustrated in Schemes 1 and 2. 1-[2-Thiazol-4-yl-(2-methylaminoethyl)]-4-n-propylpiperazine 10 (Scheme 1) was prepared from compound 5 by four-step synthesis including cyclization reaction of 1-(4-n-propyl)piperazine thioamide 5 with ethyl 4-chloroacetoacetate 6 to 1-[2-thiazol-4-yl-(2-methoxycarbonylethyl)]-4-n-propylpiperazine 7, reduction with LiAlH4 in dry ethyl ether to 1-[2-thiazol-4-yl-(2-hydroxyethyl)]-4-n-propylpiperazine 8, mesylation with methanesulfonyl chloride in dry pyridine to 1-[2-thiazol-4-yl-(2-mesyloxyethyl)]-4-n-propylpiperazine 9 and finally through nucleophilic displacement of the mesyloxy group by methylamine in methanol to 1-[2-thiazol-4-yl-(2-methylaminoethyl)]-4-n-propylpiperazine 10. 1-[2-Thiazol-4-yl-(2-methy-2-alkylaminoethyl)]-4-n-propylpiperazines 2a,b and 1-[2-thiazol-4-yl-(2-methy-2-phenylalkylaminoethyl)]-4-n-propylpiperazines 2c,d were prepared from 1-[2-thiazol-4-yl-(2-mesyloxyethyl)]-4-n-propylpiperazine 9 through nucleophilic substitution of the mesyloxy group by an appropriate secondary amine in methanol. Compounds 2e–g, 1-[2-thiazol-4-yl-(2-methyl-2-phenylalkylaminoethyl)]-4-n-propylpiperazine, were obtained from 1-[2-thiazol-4-yl-(2-methylaminoethyl)]-4-n-propylpiperazine 10 by alkylation with the corresponding primary phenyloalkyl halides in acetonitrile followed by purification with column chromatography. [2-Thiazol-4-yl-(2-metyl-2-phenylcarbonylaminoethyl)]-4-n-propylpiperazine amides 2h–k were obtained by standard methods. Compound 10 was acetylated with an appropriate acid chloride in the presence of NaHCO3 in DME, followed by purification with column chromatography.

Synthesis of 1-[2-thiazol-4-yl-(2-aminoethyl)]-4-n-propylpiperazines 2a–k

Synthesis of 1-[2-thiazol-5-yl-(2-methyl-2-phenylalkylaminoethyl)]-4-n-propyl- piperazines 3a, b and 1-[2-thiazol-5-yl-(2-methyl-2-phenylcarbonylaminoethyl)]-4-n-propyl- piperazine amides 4a–d
Compounds 3a, b, 1-[2-thiazol-5-yl-(2-methyl-2-phenylalkylaminoethyl)]-4-n-propylpiperazine (Scheme 2), were synthesized from compound 11 by alkylation with the corresponding primary phenylalkyl halides in acetonitrile followed by purification with column chromatography. Amides 4a–d were obtained by acetylation of 1-[2-thiazol-5-yl-(2-methylaminoethyl)]-4-n-propylpiperazine 11 (Scheme 2) with an appropriate acid chloride with the presence of K2CO3 in DME, followed by purification with column chromatography.
All free bases were dissolved in small amount of n-propanol and treated with methanolic HBr. The hydrobromides crystallized as white solid.
The 1-(4-n-propyl)piperazine thioamide (5) was directly obtained by the reaction of the 1-n-propylpiperazine dihydrobromide with potassium thiocyanate in aqueous solution (Frymarkiewicz and Walczynski, 2009).
The 5-phenylpentyl bromide was obtained according to Collins (Collins and Davis, 1961). The 5-phenyl-1-pentanol was converted into the bromide by treatment with 50 % aqueous hydrobromic acid and concentrated sulphuric acid.
The ethyl 4-chloroacetoacetate, 1-n-propylpiperazine dihydrobromide, benzyl bromide, 1-bromo-3-phenylpropane, 1-bromo-4-phenylbutane 5-phenyl-1-pentanol, dimethylamine solution in methanol, N-methylpropylamine, N-benzylmethylamine, N-methyl-2-phenethylamine, benzoyl chloride, p-toluoyl chloride, 4-chlorobenzoyl chloride and 4-nitrobenzoyl chloride were all purchased from commercial sources.
Results and discussion
The compounds were in vitro tested as H3 receptor antagonists—the electrically evoked contraction of the guinea-pig jejunum.
The presented series of 1-[2-thiazol-4-yl-(2-aminoethyl)]-4-n-propylpiperazines (2a–k) and their analogous 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazine (3a,b and 4a–d) derivatives possess weak to pronounced H3-receptor antagonist potency (Table 1).

The introduction of 2-methyl-2-R-aminoethyl-substituents at position 4 of the thiazole ring led to the derivatives 2a, b, d–k having, independent of the sort of substituent, weak activity, except for derivative 2c showing moderate affinity with pA2 = 7.12.
It appeared that by comparison of homologous pairs, the 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazines (3a,b and 4a–d) have much higher potency than their analogous 1-[2-thiazol-4-yl-(2-aminoethyl)]-4-n-propylpiperazines (2a–k). The differences are observed inside of each series. In the case of 1-[2-thiazol-4-yl-(2-aminoethyl)]-4-n-propylpiperazines, elongation of alkyl chain from one to three methylene groups results in an increase of potency for 2a pA2 = 6.76 and 2b pA2 = 6.96, this is in opposition to the 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazine derivatives where the 1-[2-thiazol-5-yl-(2-N,N-dimethylaminoethyl)]-4-n-propylpiperazine shows slightly higher potency than its N-methyl-N-propyl analogue (pA2 = 7.78; pA2 = 7.53, respectively).
In the 2-methyl-2-phenylalkyl derivatives of 1-[2-thiazol-4-yl-(2-aminoethyl)]-4-n-propylpiperazine (2c–g), there is no significant difference in affinity. Elongation of alkyl chain from one to five methylene groups does not influence antagonistic activity (pA2 ranging from 6.81 for compound 2d to 6.69 for compound 2g). In the analogues series, there is no significant difference in affinity among the methyl and ethyl derivatives (pA2 = 7.76 and 7.61 for compound 3a). A further elongation in the alkyl chain length to 3 methylene groups results in an increase of antagonistic activity, reaching the maximum for 1-[2-thiazol-5-yl-(2-methyl-2-phenylpropylaminoethyl)]-4-n-propylpiperazine (pA2 = 8.27); activity decreases on further lengthening up to 5 methylene groups (pA2 = 7.80 for compound 3b and 7.25 for 1-[2-thiazol-5-yl-(2-phenylpentylmethylaminoethyl)]-4-n-propylpiperazine). Replacement of hydrogen by p-benzoyl substituent at the end of N-methyl group leads to the compounds 2h–k (pA2 from 5.65 to 6.23) and their analogues 4a–d (pA2 from 7.45 to 7.76). By comparison of homologous pairs, the 1-[2-thiazol-5-yl-(2-methyl-2-phenylcarbonylaminoethyl)]-4-n-propylpiperazine amides 4a–d have much higher potency than their analogous 1-[2-thiazol-4-yl-(2-methyl-2-phenylcarbonylaminoethyl)]-4-n-propylpiperazine amides 2h–k. In both series, a slightly higher activity is observed for compounds carrying on electron-withdrawing substituent at para-position in the benzene ring.
Summarizing, 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazines display a higher activity than their 1-[2-thiazol-4-yl-(2-aminoethyl)]-4-n-propylpiperazine analogues. We observe that the position 5 of 2-methyl-2-R-aminoethyl-substituents in the thiazole ring is favourable for histamine H3 receptor antagonist activity, whereas its presence in position 4 leads, almost in each case, to strong decrease of activity.
The highest potency for both homologous series is seen in the compound with the 2-methyl-2-phenylpropylaminoethyl substituent (pA2 = 8.27) and with slightly lower potencies for compounds carrying on 2,2-dimethylaminoethyl, 2-methyl-2-(4-chlorophenyl)carbonylaminoethyl and 2-methyl-2-(4-nitrophenyl)-carbonylaminoethyl substituents (pA2 = 7.78; pA2 = 7.73 and pA2 = 7.76, respectively).
Experimental protocols
General Methods. All melting points (mp) were measured in open capillaries on an electrothermal apparatus and are uncorrected. For all compounds, 1H NMR spectra were recorded on a Varian Mercury 300 MHz spectrometer. Chemical shifts are expressed in ppm downfield from internal TMS as reference. 1H NMR data are reported in order: multiplicity (br, broad; s, singlet; d, doublet; t, triplet; m, multiplet; * exchangeable by D2O) number of protons, and approximate coupling constant in Hertz. 13C NMR spectra were recorded on Bruker Avance III 600 MHz spectrometer. Elemental analysis (C, H, N) for all compounds were measured on Perkin Elmer Series II CHNS/O Analyzer 2400 and are within ±0.4 % of the theoretical values. TLC was performed on silica gel 60 F254 plates (Merck). Flash column chromatography was carried out using silica gel 60 Å 50 μm (J. T. Baker B. V.), employing the same eluent as was indicated by TLC.
Chemistry
The synthesis of 1-[2-thiazol-4-yl-(2-methoxycarbonylethyl)]-4-n-propylpiperazine (7)
The 1-(4-n-propyl)piperazine thioamide (5) (0.032 mol) was added to a solution of ethyl 4-chloroacetoacetate (6) (0.032 mol) in 70 mL of n-propanol. The reaction mixture was heated at 90 °C for 6 h. After cooling, the solvent was removed in vacuo. The hydrochloride product was obtained as brown solid. The free base was obtained as follows: the hydrochloride of the 1-[2-thiazol-4-yl-(2-methoxycarbonylethyl)]-4-n-propylpiperazine (7) was mixed with saturated aqueous sodium bicarbonate solution for 1 h at room temperature and then water layer was extracted with dichloromethane (2 × 30 mL). The organic extracts were washed with water (3 × 30 mL), dried (Na2SO4), filtered and evaporated to give compound 7 as a sticky oil: The free base was dissolved in small amount of n-propanol and treated with methanolic HBr. The dihydrobromide crystallized as white solid.
7. C14H23N3O2S (M = 297); yield 82.6 %; sticky oil; 1H NMR (CDCl3) δ: 0.89–0.95 (t, 3H, CH2 CH 3 J = 7.5 Hz); 1.25–1.29(t, 3H, CH 3 CH2O–) 1.48–1.60 (m, 2H, –CH2 CH 2 CH3); 2.33–2.38 (m, 2H, –CH3CH2 CH 2 –); 2.52–2.56 (m, 4H CH2 CH 2 N); 3.46–3.50 (m, 4H, –CH2 CH 2 N); 3.60 (s, 2H, CH 2 CO–) 4.14–4.22(q, 2H CH 2 O, J = 7.2 Hz) 6,39 (s, 1H, H thiazole); TLC (methylene chloride:methanol 19:1) Rf = 0.21
Elemental analysis for dihydrobromide C14H25Br2N3O2 S (459.26)
C | H | N | |
|---|---|---|---|
Calculated | 36.61 % | 5.49 % | 9.15 % |
Found | 36.25 % | 5.38 % | 9.18 % |
The synthesis of 1-[2-thiazol-4-yl-(2-hydroxyethyl)]-4-n-propylpiperazine (8)
To a solution of the 1-[2-thiazol-4-yl-(2-methoxycarbonylethyl)]-4-n-propylpiperazine (7) (0.032 mol) in 110 mL of DME at 55 °C, LiBH4 (0.055 mol) was added. The mixture was stirred at 70 °C for 24 h. The solvent was evaporated and remaining material was dissolved in 60 mL of methanol and was heated at 70 °C for 24 h. The solvent was evaporated and the residue was purified by column chromatography on silica gel. The title products were obtained as sticky oil. The free base was dissolved in small amount of n-propanol and treated with methanolic HBr. The dihydrobromide crystallized as white solid.
8. C12H21N3OS (M = 256); yield 75.0 %.; 1H NMR (CDCl3) δ: 0.89–0.95 (t, 3H, CH2 CH 3 J = 7.5 Hz); 1.51–1.60 (m, 2H, –CH2 CH 2 CH3); 2.33–2.38 (m, 2H, –CH3CH2 CH 2 –); 2.52–2.56 (m, 4H CH2 CH 2 N); 2.75–2.78 (t, 2H, CH2-thiazole J = 5.7 Hz); 3.45–3.49 (m, 4H, –CH2 CH 2 N); 3.84–3.87 (t, 2H CH 2 OH, J = 5.7 Hz) 4.01 (s* br, H, OH–) 6.20 (s, 1H, H thiazole); TLC (methylen chloride:methanol 10:1) R f = 0.27.
Elemental analysis for dihydrobromide C12H21N3OSx2HBr (M = 417,22)
C | H | N | |
|---|---|---|---|
Calculated | 34.54 % | 5.56 % | 10.07 % |
Found | 34.30 % | 5.52 % | 10.07 % |
The synthesis of 1-[2-thiazol-4-yl-(2-mesyloxyethyl)]-4-n-propylpiperazine (9)
To a cooled solution of the 1-[2-thiazol-4-yl-(2-hydroxyethyl)]-4-n-propylpiperazine (8) (0.009 mol) in 10 mL of dry pyridine, while stirring, methanesulfonyl chloride (0.009 mol) was added dropwise. The mixture was stirred at room temperature for 0.5 h. Then, reaction mixture was poured out in ice-cold water (40 mL) and extracted with ethyl ether (3 × 50 mL). The combined organic extracts were dried (Na2SO4), filtered and evaporated to give compound 9 as a sticky yellow oil. The crude compound 9 was used in the next step without further purification.
9. C13H23N3O3S2 (M = 333); yield 58.1 %; 1H NMR (CDCl3) δ: 0.90–0.95 (t, 3H, CH2 CH 3 J = 7.4 Hz); 1.48–1.60 (m, 2H, –CH2 CH 2 CH3); 2.33–2.38 (m, 2H, –CH3CH2 CH 2 –); 2.52–2.56 (m, 4H CH2 CH 2 N); 2.92 (s, 3H, CH 3 SO3) 2.96–3.02 (t, 2H, CH2-thiazole J = 6.6 Hz); 3.45–3.48 (m, 4H, –CH2 CH 2 N); 4.49–4.52 (t, 2H CH 3 SO3 CH 2, J = 6.6 Hz) 6,29 (s, 1H, H thiazole); TLC (methylen chloride:methanol 10:1) Rf = 0.44.
The synthesis of 1-[2-thiazol-4-yl-(2-methylaminoethyl)]-4-n-propylpiperazine (10)
The crude 1-[2-thiazol-4-yl-(2-mesyloxyethyl)]-4-n-propylpiperazine 9 (0.008 mol) was dissolved in 30 mL of 40 % solution methylamine in methanol. The mixture was stirred at room temperature for 24 h. Then, organic solvent was evaporated, and residue was dissolved in DME (40 mL), alkalized with solid NaHCO3 (0.001 mol) and stirred for 1 h. The mixture was filtered and DME was evaporated to give compound 2 as a yellowish sticky oil. The free base was dissolved in small amount of n-propanol and treated with methanolic HBr. The treehydrobromide crystallized as white solid.
2. C13H24N4S (M = 268); yield 68.9 %; 1H NMR (CDCl3) δ: 0.90–0.95 (t, 3H, CH2 CH 3 J = 7.5 Hz); 1.50–1.60 (m, 2H, –CH 2 CH3); 2.01 (s* br, 1H, NH); 2.32–2.37 (m, 2H, –CH3CH2 CH 2 –); 2.45 (s, 3H –CH 3); 2.52–2.56; (m, 4H CH2 CH 2 N); 2.73–2.77 (t, 2H, CH 2 -thiazole, J = 6.6 Hz); 2.86–2.91 (t, 2H, CH 2N J = 6.6 Hz) 3.45–3.48 (m, 4H, CH2 CH 2 N); 6.19 (s, 1H, H thiazole); TLC (chloroform metanol concentrated ammonium hydroxide 60:10:1) Rf = 0.10.
Elemental analysis for treehydrobromide C13H27N4 Br3S (511,20)
C | H | N | |
|---|---|---|---|
Calculated | 30.54 % | 5.32 % | 10.96 % |
Found | 30.61 % | 5.23 % | 10.97 % |
General method for the preparation of 1-[2-thiazol-4-yl-(2-alkylmethylaminoethyl)] (2a,b) and 1-[2-thiazol-4-yl-(2-phenylalkylmethylaminoethyl)] 4-n-propylpiperazines (2c,d)
To a solution of 1-[2-thiazol-4-yl-(2-mesyloxyethyl)]-4-n-propylpiperazine (9) (0.002 mol) in 5.0 mL of methanol, the corresponding amine (0.004 mol) was added (in case of the compound 2a—33 % solution dimethylamine in methanol was used). The mixture was stirred at 50 °C for 6–10 h. (monitored by TLC). After the completion of reaction, the solvent was evaporated and the residue was alkalized with saturated aqueous NaHCO3 solution (15 mL) and stirred for 0.5 h. Then, the mixture was extracted with ethyl ether (3 × 30 mL). The combined organic extracts were dried (Na2SO4), filtered and evaporated. The residue was purified by column chromatography on silica gel. The title products were obtained as sticky oil. The free base was dissolved in small amount of n-propanol and treated with methanolic HBr. The hydrobromide crystallized as white solid to give compounds 2a–d.
2a. C14H26N4S (M = 282); yield 64.0 %.; 1H NMR (CDCl3) δ: 0.89–0.94 (t, 3H, –CH2 CH 3 J = 7.2 Hz); 1.47–1.57 (m, 2H, –CH2 CH 2 CH3); 2.74 (s, 3H, –NCH3); 2.31–2.36 (m, 2H, –CH3CH2 CH 2 –); 2.51–2.54 (m, 4H CH 2 CH 2 N); 2.58–2.64 (m, 2H, CH 2 N)); 2.72–2.75 (m, 2H CH2-thiazole) 3.45–3.48 (m, 4H, –CH 2 CH 2 N 6.29 (s, 1H, H thiazole); TLC (chloroform:methanol:concentrated ammonium hydroxide 40:10:1) Rf = 0.19. mpthreehydrobromide 242–244 °C.
IR (for dihydrobromide; KBr) cm−1: 3446, 3052, 2962, 2914, 2660, 2587, 2520, 2467, 1613, 1592, 1470, 1432, 1287, 1168, 1133, 997, 969, 813, 662.
Elemental analysis for dihydrobromide C14H29Br3N3S (525,22)
C | H | N | |
|---|---|---|---|
Calculated | 33.01 % | 5.57 % | 10.67 % |
Found | 32.70 % | 5.67 % | 10.62 % |
2b. C16H30N4S (M = 310); yield 68.0 %.; 1H NMR (CDCl3) δ: 0.87–0.95 (m 6H, –CH2 CH 3 ); 1.47–1.60 (m, 4H, –CH2 CH 2 CH3); 2.32 (s, 3H, –NCH 3); 2.34–2.43 (m, 4H, –CH3CH2 CH 2 –); 2.52–2.55 (m, 4H CH2 CH 2 N); 2.76 (s, 4H –NCH 2 CH 2thiazole); 3.45–3.48 (m, 4H, –CH2 CH 2 N); 6.29 (s, 1H, H thiazole); TLC (chloroform:methanol:concentrated ammonium hydroxide 40:10:1) Rf = 0.25.
IR (for treehydrobromide; KBr) cm−1: 3428, 3073, 2963, 2923, 2708, 2655, 2581, 2527, 2469, 1611, 1591, 1459, 1426,1356, 1289, 1239, 1181, 1133, 1099, 1055, 1028, 967, 898, 808, 760, 721, 638, 548.
Elemental analysis for treehydrobromide C16H33Br3N4S (553.27)
C | H | N | |
|---|---|---|---|
Calculated | 34.73 % | 6.01 % | 10.13 % |
Found | 34.71 % | 6.07 % | 10.13 % |
2c. C20H30N4S (M = 359); yield 41.0 %; 1H NMR (CDCl3) δ: 0.81–0.86 (t 3H, –CH2 CH 3 J = 7.4 Hz); 1.38–1.51 (m, 2H, –CH2 CH 2 CH3); 2.16 (s, 3H, –NCH 3); 2.22–2.28 (m, 4H, –CH3CH2 CH 2 –); 2.36–2.45 (m, 4H CH2 CH 2 N); 2.63–2.76 (m, 4H –NCH 2 CH 2-thiazole); 3.35–3.44 (m, 4H, –CH 2 CH 2 N) 3.46 (s, 2H, CH2Ph) 6.29 (s, 1H, H thiazole); 7.11–7.26 (m,5H,–H arom); TLC (chlorek metylenu:metanol 10:1) Rf = 0.23.
IR (for treehydrobromide; KBr) cm−1: 3435, 3071, 2963, 2918, 2702, 2653, 2579, 2459, 1615, 1429, 1287, 1185, 1097, 1056, 969, 751, 699.
Elemental analysis for treehydrobromide C20H33Br3N4S (601.31)
C | H | N | |
|---|---|---|---|
Calculated | 39.95 % | 5.53 % | 9.32 % |
Found | 39.57 % | 5.47 % | 9.19 % |
2d. C21H32N4S (M = 373); yield 16.9 %; 1H NMR (CDCl3) δ: 0.89–0.94 (t 3H, –CH2 CH 3 J = 7.3 Hz); 1.47–1.59 (m, 2H, –CH2 CH 2 CH3); 2.32–2.34 (m, 2H, –CH3CH2 CH 2 –); 2.36 (s, 3H, –NCH 3); 2.52–2.59 (m, 4H CH2 CH 2 N); 2.64–2.70 (m, 2H –NCH 2 CH 2-thiazole); 2.70–2.85 (m, 6H, –CH 2–thiazole –CH 2 CH 2 Ph,); 3.45–3.54 (m, 4H, –CH2 CH 2 N); 6.16 (s, 1H, H thiazole); 7.18–7.30 (m, 5H, Harom); (TLC (chloroform:metanol:amoniak 60:10:1) Rf = 0.55.
IR (for treehydrobromide; KBr) cm−1: 3430, 3071, 2962, 2928, 2702, 2653, 2577, 2458, 1613, 1594, 1456, 1411, 1357, 1289, 1181, 1098, 1055, 968, 807, 751, 698.
Elemental analysis for treehydrobromide C21H35Br3N4S (615.32)
C | H | N | |
|---|---|---|---|
Calculated | 40.72 % | 5.70 % | 9.05 % |
Found | 40.57 % | 5.37 % | 9.02 % |
General method for the preparation of 1-[2-thiazol-4-yl-(2-phenylalkylmethylaminoethyl)] 4-n-propylpiperazines (2e–g) and 1-[2-thiazol-5-yl-(2-phenylalkylmethylaminoethyl)] 4-n-propylpiperazines (3a,b)
To a solution of 1-[2-thiazol-4-yl-(2-methylaminoethyl)]-4-n-propylpiperazine (10) (0.002 mol) or 1-[2-thiazol-5-yl-(2-methylaminoethyl)]-4-n-propylpiperazine (11) (0.002 mol) with the presence of K2CO3 (0.003 mol) in 5.0 mL of acetonitrile, the corresponding phenylalkyl bromide (0.002 mol) was added. The mixture was stirred at room temperature for 6–10 h (monitored by TLC). Then, inorganic salt was filtered off and solvent was evaporated. The residue was purified by column chromatography on silica gel. The title products were obtained as sticky oil. The free base was dissolved in small amount of n-propanol and treated with methanolic HBr. The hydrobromide crystallized as white solid to give compounds 2e–g and 3a,b, respectively.
2e. C22H34N4S (M = 387); yield 39.8 %; 1H NMR (CDCl3) δ: 0.91–0.96 (t 3H, –CH2 CH 3 J = 7.3 Hz); 1.49–1.62 (m, 2H, –CH2 CH 2 CH3); 1.76–1.86 (m, 2H, –CH2 CH 2 CH2); 2.29 (s, 3H, –NCH 3); 2.33–2.38 (m, 2H, –CH3CH2 CH 2 –); 2.43–2.48 (t, 2H, –NCH 2 CH2 CH2, J = 7.5 Hz); 2.51–2.63 (m, 6H, –CH2CH2N, CH 2 Ph,); 2.71(s, 4H, –CH2-thiazole CH 2 CH 2 N); 3.42–3.45 (m, 4H, –CH2 CH 2 N); 6.34 (s, 1H, H thiazole); 7.12–7.28 (m,5H,–H arom);TLC (chloroform:metanol:amoniak 60:10:1) Rf = 0.46.
IR (for threehydrobromide; KBr) cm−1: 3428, 3075, 2962, 2922, 2649, 2577, 2519, 2458, 2363, 1620, 1453, 1430, 1403, 1286, 1240, 1185, 1134, 1033, 967, 808, 753, 700.
Elemental analysis for threehydrobromide C22H37Br3N4S (629.7)
C | H | N | |
|---|---|---|---|
Calculated | 41.98 % | 5.93 % | 8.90 % |
Found | 41.93 % | 5.96 % | 8.88 % |
2f. C23H36N4S (M = 401); yield 40.5 %; 1H NMR (CDCl3) δ: 0.90–0.94 (t 3H, –CH2 CH 3 J = 7.3 Hz); 1.47–1.67 (m, 6H, –CH2 CH 2 CH3, CH 2 CH2N; CH 2 CH2Ph); 2.27 (s, 3H, –NCH 3); 2.32–2.44 (m, 4H, –CH3CH2 CH 2 , NCH 2 CH2 CH2–); 2.41–2.49 (m, 4H CH2 CH 2 N); 2.59–2.64 (t, 2H, CH2Ph J = 7.2 Hz); 2.72 (s, 4H, –thiazole CH 2 CH 2 N); 3.42–3.48 (m, 4H, –CH2 CH 2 N); 6.16 (s, 1H, H thiazole); 7.16–7.29 (m,5H,–H arom); TLC (chloroform:metanol:amoniak 60:10:1) Rf = 0.49.
IR (for threehydrobromide; KBr) cm−1: 3523, 3422, 3067, 2965, 2938, 2705, 2655, 2582, 2529, 2469, 1613, 1592, 1457, 1413, 1357, 1289, 1182, 1097, 1029, 969, 809, 748, 705, 669, 550.
Elemental analysis for threehydrobromide C23H39Br3N4S (643.7)
C | H | N | |
|---|---|---|---|
Calculated | 42.93 % | 6.11 % | 8.71 % |
Found | 42.73 % | 6.27 % | 8.67 % |
2g. C24H38N4S (M = 415); yield 66.8 %; 1H NMR (CDCl3) δ: 0.88–0.93 (t 3H, –CH2 CH 3 J = 7.3 Hz); 1.27–1.37 (m, 2H, (CH2)2 CH 2 (CH2)2); 1.45–1.65 (m, 6H, –CH2 CH 2 CH3, CH 2 CH2N); 2.30–2.35 (m, CH3CH2 CH 2 – NCH 3); 2.41–2.52 (m, 6H, CH2 CH 2 N CH 2 CH2Ph 2.56–2.61 (t, 2H –CH 2 Ph 2,76 (s, 4H, thiazole CH 2 CH 2 N); 3.39–3.46 (m, 4H, –CH2 CH 2 N) 6.17 (s, 1H, H thiazole); 7.12–7.28 (m,5H,–H arom); TLC (chloroform:metanol:amoniak 60:10:1) Rf = 0.51.
IR (for threehydrobromide; KBr) cm−1: 3427, 3305, 3077, 2937, 2876, 2653, 2580, 2458, 1616, 1597, 1434, 1286, 1185, 1096, 967, 807, 756, 701, 528.
Elemental analysis for threehydrobromide C24H41Br3N4S (M = 657.40)
C | H | N | |
|---|---|---|---|
Calculated | 43.84 % | 6.29 % | 8.52 % |
Found | 43.75 % | 6.32 % | 8.55 % |
3a. C21H32N4S (M = 372.56); yield 48.0 %; 1H NMR (CDCl3) δ: 0.90–0.92 (t 3H. –CH2 CH 3 J = 7.2 Hz); 1.50–1.56 (m, 2H, –CH 2 CH3); 2.32–2.34 (m, 2H CH3CH2 CH 2 N); 2.35 (s, 3H CH 3 N); 2.52–2.53 (m, 4H –CH2 CH 2 N); 2.62–2.67 (m, 4H CH 2 Ph CH 2 N) 2.77–2.82 (m, 2H –CH 2 N –CH 2 -tiazol); 3.43–3.45 (m 4H –CH2 CH 2 N); 6.87 (s 1H H thiazole); 7.16–7.28 (m 5H Harom.); TLC (chloroform:methanol 9:1) Rf = 0.23.
IR (for threehydrobromide; KBr) cm−1: 3507, 3451, 3052, 2959, 2915, 2695, 2583, 2526, 1578, 1430, 1409, 1309, 1291, 1243, 1188, 1161, 1093, 1033, 964, 810, 756, 728, 703, 623, 544, 510.
Elemental analysis for threehydrobromide C21H35Br3N4S (M = 615.34)
C | H | N | |
|---|---|---|---|
Calculated | 40.99 % | 5.73 % | 9.11 % |
Found | 40.92 % | 5.51 % | 9.16 % |
3b. C23H36N4S (M = 400.62) yield 61.0 %; 1H NMR (CDCl3) δ: 0.91–0.93 (t, 3H. –CH2 CH 3 J = 7.2 Hz); 1.49–1.56 (m, 4H –CH 2 CH 2CH2N); 1.62–1.67 (m, 2H CH 2 CH3); 2.23 (s, 3H CH 3 N); 2.32–2.34 (m, 2H CH3CH2 CH 2 N); 2.38–2.40 (t, 2H J = 7.2 Hz CH 2 N); 2.50–2.55 (m, 6H –CH2 CH 2 N –CH 2 Ph); 2.61–2.63 (t, 2H J = 7.2 Hz CH 2 N); 2.77–2.79(t, 2H J = 7.2 Hz CH 2 -tiazol); 3.42–3.43 (m, 4H –CH2 CH 2 N); 6.87 (s, 1H H thiazole); 7.15–7.26 (m 5H Harom.); TLC (chloroform: methanol 9:1) Rf = 0.14.
IR (for threehydrobromide; KBr) cm−1: 3471, 3399, 3052, 2938, 2639, 2597, 2473, 1627, 1498, 1434, 1291, 1193, 1027, 964, 846, 752, 722, 597.
Elemental analysis for threehydrobromide C23H39Br3N4S (M = 643.39)
C | H | N | |
|---|---|---|---|
Calculated | 42.93 % | 6.11 % | 8.71 % |
Found | 42.87 % | 6.14 % | 8.78 % |
General method for the preparation of 1-[2-thiazol-4-yl-(2-methyl-2-phenylcarbonylaminoethyl)]-4-n-propylpiperazine amides 2h–k and 1-[2-thiazol-5-yl-(2-methyl-2-phenylcarbonylaminoethyl)]-4-n-propylpiperazine amides 4a–d
To a solution of 1-[2-thiazol-4-yl-(2-methylaminoethyl)]-4-n-propylpiperazine (2) or 1-[2-thiazol-5-yl-(2-methylaminoethyl)]-4-n-propylpiperazine (11) (0.001 mol) in 10 mL of DME, the corresponding acid chloride (0.001 mol) was added. After 15 min, NaHCO3 (0.001 mol) was added and the mixture was stirred at room temperature for 24 h. The solvent was evaporated and the residue was suspended with H2O (30 mL) and extracted with chloroform (3 × 30 mL). The combined organic extracts were dried (Na2SO4), filtered and evaporated. The residue was purified by column chromatography on silica gel. The title products were obtained as sticky oil. The free base was dissolved in small amount of n-propanol and treated with methanolic HBr. The hydrobromide crystallized as white solid to give compounds 2h–k and 4a–d, respectively. Because 1H NMR data for compounds 2h–k and 4a–d have been illegible. 13C NMR data are presented for these derivatives.
2h. C20H28N4OS (M = 372); yield 82.9 %; (δ in ppm; CDCl3, 600 MHz); 171.67; 161.18; 159.80; 137.06; 129.94; 128.00; 127.15; 122.37; 59.28; 52.05; 45.42; 43.59; 33.16; 27.08; 20.46; 13.29;. TLC (dichloromethane: methanol: 10:1) Rf = 0,36.
IR (for dihydrobromide; KBr) cm−1: 3399, 3104, 3077, 2974, 2919, 2793, 2919, 2793, 2703, 2664, 2576, 2465, 1599, 1501, 1439, 1406, 1275, 1218, 1187, 1122, 1072, 1029, 998, 967, 841, 798, 723, 637, 566, 463.
MS m/z (relative intensity) 372 (M+, 17), 274 (66), 261 (13), 152 (17), 139 (41), 126 (24), 111 (17), 105 (100), 77 (33).
Elemental analysis for dihydrobromide C20H30Br2N4OS (M = 534.37)
C | H | N | |
|---|---|---|---|
Calculated | 44.91 % | 5.28 % | 10.48 % |
Found | 45.00 % | 5.47 % | 10.58 % |
2i. C21H30N4OS (M = 386); yield 71.9 %; (δ in ppm; CDCl3, 600 MHz); 171.53; 161.18; 159.80; 139.83; 133.26; 128.69; 126.73; 121.78; 60.08; 52.05; 46.07; 44.05; 33.09; 28.34; 21.50; 20.46; 13.29;.TLC (dichloromethane: methanol: 10:1) Rf = 0.28.
IR (for dihydrobromide; KBr) cm−1: 3431, 3102, 3000, 2926, 2768, 2569, 2514, 2462, 1597, 1478, 1455, 1406, 1362, 1291, 1276, 1184, 1122, 1075, 998, 967, 834, 786, 715, 640, 565, 476.
MS m/z (relative intensity) 386 (M+, 12), 288 (43), 152 (13), 139 (22), 126 (15), 119 (100) 111 (14), 98 (20), 91 (30).
Elemental analysis for dihydrobromide C21H30Br2N4OS (M = 547.8)
C | H | N | |
|---|---|---|---|
Calculated | 46.00 % | 5.88 % | 10.22 % |
Found | 45.91 % | 5.94 % | 10.16 % |
2j. C20H27ClN4OS (M = 407); yield 49,5 %; (δ in ppm; CDCl3, 600 MHz); 171.86; 161.34; 159.80; 136.81; 132.00; 129.73; 127.53; 121.78; 59.73; 51.27; 46.95; 43.56; 31.33; 27.54; 20.46; 13.29; TLC (dichloromethane: methanol: 10:1) Rf = 0.38.
IR (for dihydrobromide; KBr) cm−1: 3101, 3072, 2967, 2928, 2759, 2706, 2574, 2463, 1617, 1596, 1441, 1408, 1291, 1215, 1186, 1122, 1093, 1073, 1014, 965, 915, 845, 786, 757, 691, 670, 639, 553, 474.
MS m/z (relative intensity) 406 (M+, 10), 308 (37), 152 (15), 141 (23), 139 (100), 126 (19), 111 (18), 98 (25).
Elemental analysis for dihydrobromide C20H29Br2ClN4OS (M = 568.81)
C | H | N | |
|---|---|---|---|
Calculated | 42.22 % | 5.14 % | 9.85 % |
Found | 42.33 % | 5.01 % | 9.98 % |
2k. C20H27N5O3S (M = 417); yield 75,5 % (δ in ppm; CDCl3, 600 MHz); 171.98; 161.57; 159.87 148.38; 143.12; 127.64; 123.71; 121.87; 55.24; 45.42; 43.81; 33.25; 27.89; 20.53; 13.32; TLC (dichloromethane: methanol: 10:1) Rf = 0.43.
IR (for dihydrobromide; KBr) cm−1: 3430, 3102, 1620, 1597, 1522, 1439, 1410, 1352, 1290, 1179, 1073, 1031, 965, 869, 851, 747, 723, 639, 558, 457.
MS m/z (relative intensity) 417 (M+, 22), 319 (100), 208 (21), 152 (32), 139 (75), 126 (26), 120 (26), 111(31), 104(31), 98 (64).
Elemental analysis for dihydrobromide C20H29Br2N5O3S (M = 579.37)
C | H | N | |
|---|---|---|---|
Calculated | 41.46 % | 5.05 % | 12.09 % |
Found | 41.45 % | 5.07 % | 12.05 % |
4a. C15H29Br3N4OS (M = 372); yield 80,1 %; (δ in ppm; CDCl3, 600 MHz); 172.87; 159.28; 138.48; 131.10; 130.04; 128.00; 126.46; 120.54; 56.47; 51.26; 45.44; 39.64; 32.76; 26.28; 20.49; 13.29;.TLC (dichloromethane:methanol: 19:1) Rf = 0.32.
IR (for dihydrobromide monohydrate; KBr) cm−1: 3509, 3436, 3046, 2971, 2923, 2681, 2586, 2522, 2464, 2084, 1629, 1607, 1575, 1443, 1402, 1360, 1294, 1221, 1098, 1075, 1023, 969, 794, 743, 714, 631, 546.
MS m/z (relative intensity) 372 (M+, 24), 274 (40), 237 (60), 224 (100), 152 (21), 139 (30), 112 (20), 105 (64), 98 (34), 77 (34).
Elemental analysis for dihydrobromide monohydrate C20H30Br2N4OS H2O (M = 552.39)
C | H | N | |
|---|---|---|---|
Calculated | 43.48 % | 5.84 % | 10.14 % |
Found | 43.73 % | 5.74 % | 10.20 % |
4b. C21H30N4OS (M = 387) yield 79,2 %; (δ in ppm; CDCl3, 600 MHz); 172.67; 159.80; 140.06; 138.48; 128.32; 125.97; 120.45; 56.39; 51.34; 45.42; 39.75; 32.84; 26.16; 21.50; 20.46; 13.29; TLC (dichloromethane: methanol: concentrated ammonium hydroxide 89:10:1) Rf = 0.51.
IR (for dihydrobromide; KBr) cm−1: 3430, 3079, 2967, 2920, 2637, 2564, 2452, 1611, 1479, 1437,1400, 1285, 1270, 1199, 1068, 1039, 968, 925, 873, 839, 757, 726, 583, 508.
MS m/z (relative intensity) 386 (M+, 20), 288 (27), 237 (80), 224 (95), 152 (25), 139 (28), 119 (100)112 (31), 111 (45), 98 (39), 91 (36).
Elemental analysis for dihydrobromide C20H30Br2N4OS (M = 534.37)
Calculated | 45.99 % | 5.88 % | 10.22 % |
Found | 45.92 % | 5.91 % | 10.16 % |
4c. C20H27ClN4OS (M = 407) yield 78,3 %; (δ in ppm; CDCl3, 600 MHz); 172.87; 159.28; 138.53; 136.18 129.26; 128.96; 127.53; 120.00; 56.39; 51.23; 45.57; 39.61; 32.82; 26.25; 20.52; 13.30; TLC (dichloromethane: methanol: concentrated ammonium hydroxide 89:10:1) Rf = 0.74
IR (for dihydrobromide; KBr) cm−1: 3522, 3422, 3034, 2988; 2938, 2896, 2656, 2569, 2458, 1622, 1430, 1399, 1339, 1291, 1257, 1174, 1089, 1039, 968, 832, 793, 758, 728, 682, 600, 552, 480.
MS m/z (relative intensity) 406 (M+, 18), 288 (27), 308 (28), 237 (34), 224 (100), 152 (64), 141 (21), 139 (92), 112 (31), 111 (43), 98 (45).
Elemental analysis for dihydrobromide C20H29Br2ClN4OS (M = 568.81)
Calculated | 42.22 % | 5.14 % | 9.85 % |
Found | 42.41 % | 5.22 % | 9.61 % |
4d. C20H27N5O3S (M = 417) yield 83.0 %; (13C δ in ppm; CDCl3, 600 MHz); 172.98; 159.67; 148.27; 140.43; 138.48; 126.87; 123.71; 120.51; 56.42; 51.56; 45.48; 39.81; 32.76; 26.22; 20.51; 13.32; TLC (dichloromethane: methanol: 10:1) Rf = 0.43.
IR (for dihydrobromide monohydrate; KBr) cm−1: 3451, 3039, 2968, 2934, 2903, 2784, 2696, 2601, 2515, 2457, 1625, 1599, 1524, 1445, 1429, 1404, 1353, 1290, 1260, 1176, 1095, 1033, 1009, 968, 870, 742, 725.
MS m/z (relative intensity) 417 (M+, 26), 319 (55), 237 (20), 224 (100), 152 (27), 150 (39) 141 (21), 139 (34),120 (25), 112 (29), 111 (68), 98 (88).
Elemental analysis for dihydrobromide monohydrate C20H29Br2N5O3S H2O (M = 597.39)
Calculated | 40.20 % | 5.23 % | 11.72 % |
Found | 40.46 % | 5.03 % | 11.77 % |
Pharmacology
All compounds were tested for H3 antagonistic effects in vitro on the guinea-pig jejunum using standard methods (Vollinga et al., 1992).
Male guinea pigs weighing 300–400 g were killed by a blow on the head. A portion of the small intestine, 20–50 cm proximal to the ileocaecal valve (jejunum), was removed and placed in Krebs buffer (composition (mM) NaCl 118; KCl 5.6; MgSO4 1.18; CaCl2 2.5; NaH2PO4 1.28; NaHCO3 25; glucose 5.5 and indomethacin (1 × 10−6 mol/L)). Whole jejunum segments (2 cm) were prepared and mounted between two platinum electrodes (4 mm apart) in 20 mL Krebs buffer, continuously gassed with 95 % O2:5 % CO2 and maintained at 37 °C. Contractions were recorded isotonically under 1.0 g tension with Hugo Sachs Hebel–Messvorsatz (Tl-2)/HF-modem (Hugo Sachs Electronik, Hugstetten, Germany) connected to a pen recorder. After equilibration for 1 h with every 10 min washings, the muscle segments were stimulated maximally between 15 and 20 V and continuously at a frequency of 0.1 Hz and a duration of 0.5 ms, with rectangular-wave electrical pulses, delivered by a Grass Stimulator S-88 (Grass Instruments Co., Quincy, USA). After 30 min of stimulation, 5 min before adding (R)-α-methylhistamine, pyrilamine (1 × 10−5 mol/L concentration in organ bath) was added, and then cumulative concentration–response curves (half-log increments) of (R)-α-methylhistamine, H3-agonist were recorded until no further change in response was found. Five minutes before adding the tested compounds, the pyrilamine (1 × 10−5 mol/L concentration in organ bath) was added, and after 20 min cumulative concentration–response curves (half-log increments) of (R)-α-methylhistamine, H3-agonist, were recorded until no further change in response was found. Statistical analysis was carried out with the Students’ t test. In all tests, p < 0.05 was considered statistically significant. The potency of an antagonist is expressed by its pA2 value calculated from the Schild (Arunlakshana and Schild, 1959) regression analysis where at least three concentrations were used. The pA2 values were compared with the potency of thioperamide.
References
Apodaca R, Dvorak CA, Xiao W, Barbier AJ, Boggs JD, Wilson SJ, Lovenberg TW, Carruthers NIA (2003) A new class of diamine-based human histamine H3 receptor antagonists: 4-(aminoalkoxy)benzylamines. J Med Chem 46:3938–3944
Arrang J-M, Garbarg M, Schwartz J-C (1983) Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature (London) 302:832–837
Arrang J-M, Garbarg M, Schwartz J-C (1987) Autoinhibition of histamine synthesis mediated by presynaptic H3-receptors. Neuroscience 23:149–157
Arunlakshana O, Schild HO (1959) Some quantitative uses of drug antagonists. Br J Pharmacol 14:48–55
Brimblecombe RW, Duncan WA, Durant GJ, Emmett JC, Ganellin CR, Leslie GB, Parsons ME (1978) Characterization and development of cimetidine as a histamine H2-receptor antagonist. Gastroenterology 74:339–347
Brown RE, Reymann KG (1996) Histamine H3 receptor-mediated depression of synaptic transmission in the dentate gyrus of the rat in vitro. J Physiol 496:175–184
Celanire S, Wijtmans M, Talaga P, Leurs R, de Esch IJP (2005) Keynote review: histamine H3 receptor antagonists reach out for the clinic. Drug Discov Today 10:1613–1627
Cemkov MJ, Davenport AJ, Harich S, Ellenbroek BA, Cesura A, Hallett A (2009) The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov Today 14:509–515
Clapham J, Kilpatrick GJ (1992) Histamine H3 receptors modulate the release of [3H]-acetylcholine from slices of rat entorhinal cortex: evidence for the possible existence of H3 receptor subtypes. Brit J Pharmacol 107:919–923
Collins RF, Davis M (1961) The chemotherapy of schistosomiasis. Part IV. Some ethers of 4-amino-2-methoxyphenol. J Chem Soc 1863–1879
Cowart MD, Bennani YL, Faghih R, Gfesser G (2002) Novel amnies as histamine-3 receptor ligands and their therapeutic applications (Abbott Laboratories). WO 02074758. [Chem. Abstr. 137 (2002) P247602x].
Cowart M, Altenbach R, Black L, Faghih R, Zhao C, Hanckok AA (2004) Medicinal chemistry and biological properties of non-imidazole histamine H3 antagonists. Mini-Rev Med Chem 4:979–992
Esbenshade TA, Fox CB, Cowart MD (2006) Histamine H3 receptor antagonists: preclinical promise for treating obesity and cognitive disorders. Mol Interv 6:77–88
Frymarkiewicz A, Walczyński K (2009) Non-imidazole histamine H3 ligands, part IV: SAR of 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazine derivatives. Eur J Med Chem 44:1674–1681
Ganellin CR, Lurquin F, Piripitsi A, Arrang J-M, Garbarg M, Ligneau X, Schunack W, Schwartz J-C (1998) Synthesis of potent non-imidazole histamine H3-receptor antagonists. Arch Pharm Med Chem 331:395–404
Hancock AA (2003) H3 receptor antagonists/inverse agonists as anti-obesity agents. Curr Opin Invest Drugs 60:25–28
Hancock AA, Busch EN, Jacobson PB, Faghih R, Esbenshade TA (2004) Histamine H(3) antagonists in models of obesity. Inflamm Res 53:547–548
Hough LB (2001) Genomics meets histamine receptors: new subtypes, new receptors. Mol Pharmacol 59:415–419
Leurs R, Church MK, Taglialatela M (2002) H1-antihistamines: inverse agonism, anti-inflammatory actions and cardiac effects. Clin Exp Allergy 32(4):489–498
Lin JH, Lu AYHI (1998) Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet 35:361–390
Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, Jackson MR, Erlander MG (1999) Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol 55:1101–1107
Meier G, Apelt J, Reichert U, Grassman S, Ligneau X, Elz S, Leurguin F, Ganellin CR, Shwartz J-C, Schunack W, Stark H (2001) Influence of imidazole replacement in different structural classes of histamine H(3)-receptor antagonists. Eur J Pharm Sci 13:249–259
Monti JM (1993) Involvement of histamine in the control of the waking state. Life Sci 53:1331–1338
Quades RD (1987) Attention deficit disorder with hyperactivity. (ADDH): the contribution of catecholaminergic activity. Prog Neurobiol 29:365–391
Schlicker E, Betz R, Göthert M (1988) Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn-Schmiedeberg’s Arch Pharmacol 337:588–590
Schlicker E, Schunack W, Göthert M (1990) Histamine H3 receptor-mediated inhibition of noradrenaline release in pig retina discs. Naunyn-Schmiedeberg’s Arch Pharmacol 342:497–501
Schlicker E, Fink K, Detzner M, Göthert M (1993) Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J Neural Transm Gen Sect 93:1–10
Stark H, Schlicker E, Schunack W (1996) Developments of histamine H3-receptor antagonists. Drug Futur 21(5):507–520
Thurmond RL, Desai PJ, Dunford PJ, Fung-Leung WP, Hofstra CL, Jiang W, Nguyen S, Riley JP, Sun S, Williams KN, Edwards JP, Karlsson LJ (2004) A potent and selective histamine H4 receptor antagonist with anti-inflammatory properties. J Phaemacol Exp Ther 309:404–413
Vahora D, Pal SN, Pillai KK (2001) Histamine and selective H3-receptor ligands: a possible role in the mechanism and management of epilepsy. Pharmacol Biochem Behav 68:735–741
Van der Goot H, Timmerman H (2000) Selective ligands as tools to study histamine receptors. Eur J Med Chem 35:5–20
Velligan DI, Miller AL (1999) Cognitive dysfunction in schizophrenia and its importance to outcome: the place of atypical antipsychotics in treatment. J Clin Psychiatry 60:25–28
Vollinga RC, Zuiderveld OP, Scheerens H, Bast A, Timmerman H (1992) A simple and rapid in vitro test system for the screening of histamine H3 ligands. Methods Find Exp Clin Pharmacol 105:747–751
Walczyński K, Guryn R, Zuiderveld OP, Timmerman H (1999) Non-imidazole histamine H3 ligands, part 2: new 2-substituted benzothiazoles as histamine H3 antagonists. Arch Pharm Pharm Med Chem 332:389–398
Walczyński K, Zuiderveld OP, Timmerman H (2005) Non-imidazole histamine H3 ligands. Part III. New 4-n-propylpiperazines as non-imidazole histamine H3-antagonists. Eur J Med Chem 40:15–23
Yokatoni K, Murakami Y, Okada S, Wang M, Nakamura K (2000) Histamine H(3) receptor-mediated inhibition of endogenous acetylcholine release from the isolated, vascularly perfused rat stomach. Eur J Pharmacol 392:23–29
Zhang M, Ballard ME, Pan P, Roberts S, Faghih R, Cowart MD, Esbenshade TA, Fox G, Decker MW, Hancock AA, Rueter LE (2005) Lack of cataleptogenic potentiation with non-imidazole H3 receptor antagonists reveals potential drug–drug interactions between imidazole-based H3 receptor antagonists and antipsychotic drugs. Brain Res 1045:142–149
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Guryn, R., Staszewski, M. & Walczyński, K. Non-imidazole histamine H3 ligands: part V. synthesis and preliminary pharmacological investigation of 1-[2-thiazol-4-yl- and 1-[2-thiazol-5-yl-(2-aminoethyl)]-4-n-propylpiperazine derivatives. Med Chem Res 22, 3640–3652 (2013). https://doi.org/10.1007/s00044-012-0372-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-012-0372-8