
Abstract
Correlative light and electron microscopy is a powerful technique for identification and determination of the structures of interested macromolecules in situ. Combined with sample vitrification, it would be much easier to preserve the native state of macromolecule complexes and distinguish them from the crowded structure environment. In this article, we present a detailed process for the application of the CorrSight system, a light microscope equipped with a cryo module, in combination with a cryo-electron microscope. A relatively long course of up to 7–8 h for cryo module preparation and multichannel light microscopy imaging of vitrified specimen can be sustained. Correlation of light and electron microscopy images at both grid levels to locate squares and square level to locate target particles, and verification of target particles can be performed with the help of AutoEMation software. Cryo-electron tomography is used for obtaining the three-dimensional structure information.
Similar content being viewed by others

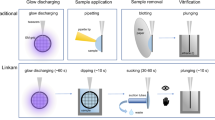

Introduction
Vitrification of biological specimens in liquid nitrogen (LN2) temperature has been proved to be a powerful technique to preserve the native structures of macromolecules either in vitro or in situ. In combination with the most recent hardware and software breakthroughs, cryo-fixed macromolecule complexes isolated from cells can be determined at near atomic resolution using the single particle reconstruction method (Ma et al. 2017; Peng et al. 2016; Wang 2015; Wang et al. 2017a). However, it is still very challenging to identify and determine the structures of interested macromolecules in situ with cryo-electron microscopy (cryo-EM) (Lucic et al. 2008; Oikonomou and Jensen 2017; Plitzko et al. 2009). The major hurdle lies in the fact that many different kinds of macromolecules are crowded within the cell and therefore are hard to be distinguished from each other simply by their shapes revealed in cryo-EM (Bauerlein et al. 2017; Zhang 2013). Furthermore, the large dimension of a cell and a rather small viewing area of EM at a high magnification render it more difficult to localize relatively sparse molecules in a specific cellular structure environment (Plitzko et al. 2009). Light microscopy (LM), more specifically, fluorescence microscopy (FM) is well developed to label specific molecules and localize them in a large viewing area with a resolution of several hundreds of nanometers. The advantages of specific labeling and localization in LM and high spatial resolution in EM can be combined, leading to the development of a powerful technique as the correlative light and electron microscopy (CLEM) (Anderson et al. 2017; Compera et al. 2015; Faas et al. 2013; Koning et al. 2014; Sjollema et al. 2012).
CLEM technique was first reported in the early 1960s for adenovirus study with conventional chemical fixation EM specimen preparation (Godman et al. 1960; Morgan et al. 1960). While the chemical fixed specimens are still the majority of targets investigated by CLEM (Hellstrom et al. 2015; Kobayashi et al. 2016; Kong and Loncarek 2015; Loussert Fonta and Humbel 2015; Mourik et al. 2014), vitrified biological specimens become more and more popular to avoid the potential artifacts and damages caused by the chemical fixation. Cryo-CLEM is thus developed in order to visualize vitrified specimens by cryo-EM (Bykov et al. 2016; Mahamid et al. 2016; Wolff et al. 2016). Currently, two types of methods are used for correlation: (1) correlative LM/EM with freezing after FM imaging (Briegel et al. 2010). FM imaging of samples at room temperature could be done with any typical FM instrument. FM can have better resolution with high-powered, oil-immersion lenses with large numerical apertures. After FM imaging, the sample needs to be frozen for further cryo-EM imaging. Various factors can be introduced to the specimen between the LM imaging and specimen vitrification, causing more difficulties for accurate correlation. Therefore, it works better for specimen that is relatively stable and the fluorescently marked structure would not change in the seconds to minutes before vitrification. (2) Correlative LM/EM with freezing before FM imaging (Briegel et al. 2010). The sample is plunge frozen or cryo-sectioned first, then the grid is visualized under LM and EM sequentially with little disturbance of its structure, thus making the correlation more consistent. Such a practice needs the cryo-fixed specimen to be maintained always in a humidity-free environment and below ~−140 °C during the whole CLEM process to prevent ice contamination on the specimen surface or recrystallization in the specimen. Special specimen stages designed for the related instruments are essential to preserve the cryo-fixed specimens (Li et al. 2018; Liu et al. 2015; Plitzko et al. 2009; Rigort et al. 2010; Schorb and Briggs 2014; Schorb et al. 2017; Zhang 2013). While the first type of CLEM method provides convenience of use and better resolution of LM, the second type guarantees more accurate correlation and is more favored (Briegel et al. 2010).
For the second type of CLEM method, two different designs are commercially available. One is the integrated (Agronskaia et al. 2008; Faas et al. 2013) and the other is the separated. The first integrated CLEM system called Tecnai with iCorr (Thermo Fisher Scientific Inc.) allows the stage to be tilted 90° to switch between the LM and EM mode for direct correlation of a specimen in situ, preventing the specimen from potential contamination and damage. The experiments efficiency is greatly increased without repeated grid transfers (Agronskaia et al. 2008). Within the iCorr system, however, only small light objective lens as 15×/0.5 NA can be integrated due to the limited space between the pole pieces of a standard Tecnai Spirit TEM. The iCorr system thus can only have fluorescence signals at the green channel with a low resolution of about 500 nm (Wang et al. 2017b). In comparison, separated CLEM systems such as the CorrSight system equipped with a cryo module from Thermo Fisher Scientific Inc. (Arnold et al. 2016) and Leica cryo-CLEM System (Hampton et al. 2017) allow the observation of a cryo-fixed specimen in a more advanced LM at better resolution and in an EM with high acceleration voltage. Different image processing software packages have been developed for high-precision correlation. The study of virus-infected or transfected mammalian cells by using Leica Cryo system has been reported. Here we present a detailed process for the application of the CorrSight system in combination with a cryo-TEM. We used plunge-frozen technique to fix the biological specimen and cryo-electron tomography (cryo-ET) for obtaining the three-dimensional (3D) structure information.
Experiment procedures
Sample preparation
In our CLEM study, biological samples were vitrified and assembled into EM Autogrids as below:
-
(1)
GiG R3.5/1 200 mesh grids (GiG C200F1, Changshu Zhongke Xinghua Technology) with indexed letters and numbers were glow discharged by means of a basic plasma cleaner PDC-32G-2 (Harrick Plasma).
-
(2)
After biological sample was applied onto the grids, cryo-EM grids were prepared using Vitrobot Mark IV (Thermo Fisher Scientific Inc.) at 20 °C and with 100 percent humidity. The blot force is set as −2 and the blot time is set as 2–2.5 s. The vitrified specimen on cryo-EM grids was stored in LN2 dewars for further examination.
-
(3)
The assembly of Autogrids is executed inside a regular Cryo Transfer Workstation (Thermo Fisher Scientific Inc.) for a TEM with autoloader. Each grid is placed inside a C-clip ring (Thermo Fisher Scientific Inc.) with the sample-application side facing down and then clipped with a C-clip (Thermo Fisher Scientific Inc.) from the top to form a sandwich-type Autogrid.
-
(4)
In the cryo shuttle and Transfer Box Assembly Workstation (Thermo Fisher Scientific Inc.) of the CorrSight system, the Autogrids are mounted on a cryo shuttle with the C-clip ring side facing up (Fig. 1). The metal spacing cylinder is then put on top of the Autogrids, and the Plexiglas lid is closed immediately. Two Autogrids can be loaded into one cryo shuttle. The cryo shuttle can be stored in the transfer box.
Fig. 1 
Cryo shuttle and transfer box assembly for CorrSight. A The display of cryo shuttle components and transfer box. Lid and cylinders are used for fixing Autogrids. B–D The process of assembly. Cryo shuttle is assembled after mounting Autogrids on cryo shuttle (1), putting cylinders (2) and then putting lid (3) on the grids as the numbers correspond to the sequence illustrated in B. The assembled cryo shuttle in C is eventually loaded into transfer box in D

The steps (2)–(4) should all be performed at LN2 temperature. The Autogrids or transfer boxes are all stored in LN2 dewars for further cryo-LM or cryo-EM imaging.
Fluorescence imaging with the CorrSight system
The flowchart of fluorescence imaging with the CorrSight system is illustrated as shown in Fig. 2. Before LM imaging of cryo samples, purging of the objective (chamber) with dry nitrogen gas and cooling down of the cryo module with liquid nitrogen are processed for a total time of 2–3 h (Fig. 3). Because of the continuous objective purging demand during the whole process, a pipeline supply of dry nitrogen gas is preferred or a self-pressurized LN2 dewar is the second choice. In this experiment, a few dry nitrogen gas cylinders were used, which can sustain as long as 8 h, from the objective purging to system heating. High-pressure gas purging on the cover glass must be done for de-icing, after the temperature of sample and chamber reaches a range from −185 to −195 °C (Fig. 4C). Live imaging mode of the system can be used to check whether the cover glass is clean enough for light source to penetrate, especially at the location of the grids. When the system is stable, the transfer box can be transferred into the cryo module chamber in a far end, and the cryo shuttle is placed in the center of the bottom, with the screw facing toward the operator (Fig. 3B). The fluorescence imaging steps are detailed as follows:

Flowchart of fluorescence imaging with CorrSight

The use of the cryo module of CorrSight. A Purging and cooling of the cryo module chamber before LM imaging. B The overhead view of cryo module chamber. There are locations with similar shape of transfer box and cryo shuttle. The screw of cryo shuttle should be facing toward the user. The right sample numbered 1 would be viewed as the left one in Maps software, while the left sample numbered 2 would be viewed as the right one in Maps software. C Warm up of the cryo module chamber at 65 °C with the heater and nitrogen gas injection after LM imaging

The acquisition of multichannel LM images with Maps software. A The samples 1 and 2 correspond to the ones described in Fig. 3B. B Layer of sample 1. Each sample can have a layer which may contain one low magnification grid map and many high-magnification fluorescence square images numbered in sequence. C The temperature of sample and chamber should be kept in the range from −185 to −195 °C during imaging process
-
(1)
Check the intactness of carbon film, flatness of grid, and thickness of vitreous ice for the Autogrids (Fig. 4A), using the 5×/0.16 NA or 10×/0.3 NA objective lenses. Fluorescence signal can be captured with the 20×/0.8 NA or 40×/0.9 NA objective lenses. Screen the cryo grids to choose the best one (called finder grid) for further image acquisition.
-
(2)
Acquire the montaged map of the finder grid at a magnification 5×/0.16 NA or 10×/0.3 NA with the cryo trans-illumination channel and autofocus strategy (Fig. 5A). Stitch tiles to minimize the gap.
Fig. 5 
The correlation of LM and EM grid map. A The montaged LM trans-illumination image of the grid at a magnification of 5×/0.16 NA. More than 60 squares are multichannel imaged and labeled with sequence number, which corresponds to the number in Fig. 4B. B The montaged EM image of the grid at a nominal magnification of 40×. The squares with targeted fluorescence signal are marked after the correlation of LM and EM grid map with AutoEMation
-
(3)
Multichannel images of interested squares can be acquired at a magnification of 40×/0.9 NA as exampled in Fig. 6 shows clear fluorescence signals. Acquire images with z-stack mode if desired. Wide-field or spinning disk acquisition mode can be chosen according to the fluorescence intensity, the imaging resolution, and so on. For Wide field fluorescence, there is Xe-lamp light source and the oligochrome has three lines (405, 488, and 561 nm). For spinning disk confocal, four laser line combiner (405, 488, 561, and 640 nm) is provided. The Maps software (Thermo Fisher Scientific Inc.) provided with CorrSight allows the z-stack MIP (maximum intensity projection) averaging and adjustment of the correlation parameters, such as the contrast and transparency. Overlay of the transillumination and fluorescence images make available the identified information for EM correlation. Adjust the fine alignment between 5×/0.16 NA and 40×/0.9 NA images manually to locate good squares within the finder grid. This alignment can be done only once at the beginning, unless the cryo shuttle is moved. Good squares are normally the ones coated with a complete layer of carbon film and free from indexed letters and numbers as shown in Fig. 5A. Note that during the process of fluorescence imaging, the cover glass should be purged repeatedly to prevent ice growth, especially in a humid environment. Every time during the purging, the cryo shuttle should be put back into the transfer box to protect grids from temperature increase during the purging. Keep live transillumination imaging by focusing on the cover glass, the crystal ice would be blown away gradually. The temperature changes of the samples and specimen chambers should be closely monitored, especially after continuous imaging for more than 2 h, to ensure that all liquid nitrogen in the cryo module dewar is not used up (Fig. 3A).
Fig. 6 
A typical LM image of a square at a magnification of 40×/0.9 NA showing clear fluorescence signals
Note: Turn on the air compressor only at the beginning of the multichannel image acquisition and turn it off before the cryo sample transfer to prevent severe vibration of CorrSight. Once the imaging is completed, the cryo samples should be unloaded and put back in the LN2 tank immediately.
-
(4)
After the image acquisition, put the cryo shuttle back to the LN2 storage tank. The cryo module should be heated continuously at 65 °C for 2 h and purged at 0.5 bar with pressurized LN2 tank at the same time (Fig. 3C).
-
(5)
Overlaid multichannel images can be adjusted and saved manually for EM correlation. Within a good grid, more than 60 squares could be imaged and marked (Figs. 4B, 5A). This is the step for rough and quick screening of squares. Find squares with interested fluorescence signal and label the locations within the overlaid images (Fig. 5A) for further cryo-EM imaging.


Correlation of cryo-LM and cryo-EM images
Two functions in the upgraded version 2.0 of AutoEMation software (Lei and Frank 2005) are used for acquisition of montaged EM atlas (Fig. 7A), correlation of LM and EM images (Fig. 7B).

Two functions in AutoEMation used for this CLEM study. A Acquisition of montaged EM atlas of grid and square. B Correlation of LM and EM images
To locate and verify target particles for cryo-ET data collection, there are three sequential steps described as follows:
-
(1)
The first step is the correlation of LM and EM grid map to locate the selected squares in EM grid map.
After loading the finder grid from the cryo shuttle into a cryo transmission electron microscope, for example, a Cs-corrected Titan Krios in this work, by using the Cryo Transfer Workstation, a montaged EM atlas of a grid is acquired at a nominal magnification in low magnification range (40× for Gatan Orius SC200 CCD camera, 100× for Falcon camera, 175× for Gatan GIF K2 camera) as exampled in Fig. 5B. The acquisition usually starts from the stage center and stage movement is used.
The conversion matrix between the LM and EM grid maps should be determined, which can be derived from two reference positions, i.e., indexed letters and numbers, recognizable in both the LM and EM maps. For each reference position, the coordinate in the LM map is obtained by using any graphic software, while the coordinate in the EM map is obtained and converted to the corresponding EM stage position via a precalibrated matrix between the camera and stage by using AutoEMation.
With the help of the conversion matrix, the coordinates of those marked squares in the LM map (Fig. 5A) can be converted to their corresponding coordinates in the EM map marked (Fig. 5B) by clicking the “Mark” button in Fig. 7B, and the EM stage positions which can be reached by clicking the “GoTo” button near to the “Mark” button directly or the “Go to XYZ” button (Fig. 7B) if the marker in the EM map is selected (Fig. 5B).
-
(2)
The second step is the correlation of LM and EM square map to locate target particles in EM square map.
For each marked square, a montaged EM atlas of the square (Fig. 8C) is acquired at the lowest nominal magnification in the SA range (1700× for both Gatan Orius SC200 CCD camera and Falcon camera, 2250× for Gatan GIF K2 camera). The acquisition starts locally after reaching the stage position of the square in the above step and stage movement or a combination of stage movement and image-beam shift is used.

The correlation of LM and EM square images and target identification. A The LM image of a square at a magnification of 40×/0.9NA. B Zooming in on the area highlighted in A. Three vesicles can be identified and marked by asterisks in red, blue, and yellow, respectively, which are close to the fluorescence signal, highlighted by yellow circle. C The montaged EM image of a square at a nominal magnification of 1700×. D Zooming in on the area highlighted in C. The target can be located via correlation by means of AutoEMation and verified with the three neighboring vesicles identified accordingly. E Low-dose EM imaging for verification of the target at a nominal magnification of 18,000×. F Tomogram slice of the target after tomographic data collection and reconstruction
Similarly, the conversion matrix between the LM and EM square maps should be determined, which can be derived from two reference positions, i.e., square corners or any features, recognizable in both the LM and EM maps.
With the help of the conversion matrix, the coordinates of fluorescence target in the LM map (Fig. 8A, B) can be converted to their corresponding coordinates in the EM map (Fig. 8C, D) marked by clicking the “Mark” button in Fig. 7B, and the EM stage positions which can be reached by clicking the “GoTo” button near to the “Mark” button directly or the “Go to XYZ” button (Fig. 7B) if the marker in the EM map is selected. The location of the target particle can be finely tuned with the combination of “Refresh” and “Go to XY” buttons (Fig. 7B) as the low-dose alignment between this magnification and a much higher magnification for verification can be set precisely with the inaccuracy of less than 0.02 μm by using AutoEMation.
Sometimes it is useful to verify the target particle visually. In Fig. 8B and D, three vesicles near the fluorescence signal are used as references for verification.
-
(3)
The third step is the verification of the target particles at a much higher magnification to ensure they are suitable for cryo-ET data collection.
The target particles selected from the above two steps may not be suitable for cryo-ET data collection as images acquired at the above magnification cannot provide enough details. Therefore, it is worth imaging the target particles at a much higher magnification (i.e., nominal magnification of 18,000× for Falcon camera, 2–3 e/Å2 dose, −8 to −10 μm defocus) for verification as further cryo-ET data collection takes very long time. The imaging for verification can be simply done by clicking the “Refresh” button (Fig. 7B) after switching to the preset high-magnification mode.
Cryo-ET data collection and reconstruction
Cryo-ET data collection and reconstruction is usually performed by using free software packages and/or packages from EM vendors.
Set batch tomography data collection positions for all targets
-
(1)
Set up FEI TEM tomography and build a new specimen session by checking the low-dose and batch options. Select file format and output folder.
-
(2)
Add every target with separate defocus settings, ranging from −5 to −8 μm. Set the focus and tracking areas away from the exposure area. Determine the exposure time of data collection based on the limit of 1–2 e/Å2 for each tilted image.
-
(3)
As a typical setting, acquisition usually starts at 0° and single-axis tilt series are collected with 1.5° increment between −65° and 65° for frozen samples. Autofocus is performed before image acquisition periodically. The general periodicity switch angle is 30° as focus would be checked every five images when the tilt angle is lower than 30° and every two images when the tilt angle is higher than 30°. In order to track the targets during the tilting, “tracking before” is usually performed when the tilt angle is lower than 10° and “tracking after” is performed for other tilt angles.
Data alignment and reconstruction
For the tomographic data with fiducial gold, use IMOD software package to do alignment and reconstruction. Execute the Protomo software package (Winkler and Taylor 2006) for the alignment and reconstruction for tomographic data without gold particles. Low-pass and high-pass filtering might be used for the noisy images, which is particularly helpful for the cross-correlation calculation during alignment. With Protomo, four or five iterations is enough for the 2 k × 2 k images and the weighted back-projection algorithm is used to generate the final 3D reconstruction from raw images. Apply median filtering or nonliner anisotropic diffusion filtering distributed in IMOD to enhance the contrast and signal-to-noise ratio of the tomograms. Further analysis of subvolume averaging and segmentation can be performed in Dynamo and Amira, respectively.
Discussion
The CorrSight system can sustain cryo-LM imaging for a long duration of up to 5 h. The Maps software can save all the acquired data automatically in the project and avoid data loss due to unexpected interruption such as software crashing. More importantly, the packed Autogrids matches well with cryo-SEM, cryo-TEM with autoloader, and Cryo Transfer Workstation. The application of AutoEMation software makes the correlation much easier. For the manual verification during the correlation, fluorescent beads can be used if there are no distinguishable particles. Verification with high-magnification imaging is necessary before cryo-ET data collection although the target is preexposed with a small amount of dose, as acquisition of each tilt series would take nearly 1 h.
The protocol in this study applies mainly to relatively thin samples, which can be prepared by plunge freezing by Vitrobot or similar apparatus and imaged with cryo-TEM directly. If the samples are too thick for electron beam to penetrate, ultrathin sectioning under LN2 environment needs to be executed before the sample can be loaded into the cryo-TEM. Sometimes high-pressure freezing should be performed instead of fast frozen for large cells or tissues, to get vitreous ice for the whole volume (Mahamid et al. 2015).
Besides the cryo- sectioning by diamond knife (Cortese et al. 2013), milling by cryo-focused ion beam (cryo-FIB) recently becomes the priority to get artifact-free, thin, frozen-hydrated lamella via fabrication (Arnold et al. 2016; Beck and Baumeister 2016; Lucic et al. 2013). After cryo-LM imaging, the locations with fluorescence signal can be specifically milled by FIB to generate the thin lamella, followed by cryo-EM high-resolution data collection (Fukuda et al. 2014). In addition, the resolution of LM imaging needs to be improved by integrating a super-resolution LM platform and a well-designed cryo module, which would be very powerful for the correlation of light and electron microscopy. In the future, more integrated and automated system to bring all the above-mentioned steps in a seamless pipeline would make the correlative cryo-EM more powerful and robust.
References
Agronskaia AV, Valentijn JA, van Driel LF, Schneijdenberg CT, Humbel BM, van BergenHenegouwen PM, Verkleij AJ, Koster AJ, Gerritsen HC (2008) Integrated fluorescence and transmission electron microscopy. J Struct Biol 164:183–189
Anderson KL, Page C, Swift MF, Hanein D, Volkmann N (2017) Marker-free method for accurate alignment between correlated light, cryo-light, and electron cryo-microscopy data using sample support features. J Struct Biol 201:46–51
Arnold J, Mahamid J, Lucic V, de Marco A, Fernandez JJ, Laugks T, Mayer T, Hyman AA, Baumeister W, Plitzko JM (2016) Site-specific cryo-focused ion beam sample preparation guided by 3D correlative microscopy. Biophys J 110:860–869
Bauerlein FJB, Saha I, Mishra A, Kalemanov M, Martinez-Sanchez A, Klein R, Dudanova I, Hipp MS, Hartl FU, Baumeister W, Fernández-Busnadiego R (2017) In situ architecture and cellular interactions of PolyQ inclusions. Cell 171(179–187):e110
Beck M, Baumeister W (2016) Cryo-electron tomography: can it reveal the molecular sociology of cells in atomic detail? Trends Cell Biol 26:825–837
Briegel A, Chen S, Koster AJ, Plitzko JM, Schwartz CL, Jensen GJ (2010) Correlated light and electron cryo-microscopy. Methods Enzymol 481:317–341
Bykov YS, Cortese M, Briggs JA, Bartenschlager R (2016) Correlative light and electron microscopy methods for the study of virus–cell interactions. FEBS Lett 590:1877–1895
Compera D, Entchev E, Haritoglou C, Mayer WJ, Hagenau F, Ziada J, Kampik A, Schumann RG (2015) Correlative microscopy of lamellar hole-associated epiretinal proliferation. J Ophthalmol 2015:450212
Cortese K, Vicidomini G, Gagliani MC, Boccacci P, Diaspro A, Tacchetti C (2013) High data output method for 3-D correlative light-electron microscopy using ultrathin cryosections. Methods Mol Biol 950:417–437
Faas FG, Barcena M, Agronskaia AV, Gerritsen HC, Moscicka KB, Diebolder CA, van Driel LF, Limpens RW, Bos E, Ravelli RB, Koning RI, Koster AJ (2013) Localization of fluorescently labeled structures in frozen-hydrated samples using integrated light electron microscopy. J Struct Biol 181:283–290
Fukuda Y, Schrod N, Schaffer M, Feng LR, Baumeister W, Lucic V (2014) Coordinate transformation based cryo-correlative methods for electron tomography and focused ion beam milling. Ultramicroscopy 143:15–23
Godman GC, Morgn C, Breitenfeld PM, Rose HM (1960) A correlative study by electron and light microscopy of the development of type 5 adenovirus. II. Light microscopy. J Exp Med 112:383–402
Hampton CM, Strauss JD, Ke Z, Dillard RS, Hammonds JE, Alonas E, Desai TM, Marin M, Storms RE, Leon F, Melikyan GB, Santangelo PJ, Spearman PW, Wright ER (2017) Correlated fluorescence microscopy and cryo-electron tomography of virus-infected or transfected mammalian cells. Nat Protoc 12:150–167
Hellstrom K, Vihinen H, Kallio K, Jokitalo E, Ahola T (2015) Correlative light and electron microscopy enables viral replication studies at the ultrastructural level. Methods 90:49–56
Kobayashi S, Iwamoto M, Haraguchi T (2016) Live correlative light-electron microscopy to observe molecular dynamics in high resolution. Microscopy 65:296–308
Kong D, Loncarek J (2015) Correlative light and electron microscopy analysis of the centrosome: a step-by-step protocol. Methods Cell Biol 129:1–18
Koning RI, Celler K, Willemse J, Bos E, van Wezel GP, Koster AJ (2014) Correlative cryo-fluorescence light microscopy and cryo-electron tomography of Streptomyces. Methods Cell Biol 124:217–239
Lei J, Frank J (2005) Automated acquisition of cryo-electron micrographs for single particle reconstruction on an FEI Tecnai electron microscope. J Struct Biol 150:69–80
Li S, Ji G, Shi Y, Klausen LH, Niu T, Wang S, Huang X, Ding W, Zhang X, Dong M, Xu W, Sun F (2018) High-vacuum optical platform for cryo-CLEM (HOPE): a new solution for non-integrated multiscale correlative light and electron microscopy. J Struct Biol 201:63–75
Liu B, Xue Y, Zhao W, Chen Y, Fan C, Gu L, Zhang Y, Zhang X, Sun L, Huang X, Ding W, Sun F, Ji W, Xu T (2015) Three-dimensional super-resolution protein localization correlated with vitrified cellular context. Sci Rep 5:13017
Loussert Fonta C, Humbel BM (2015) Correlative microscopy. Arch Biochem Biophys 581:98–110
Lucic V, Leis A, Baumeister W (2008) Cryo-electron tomography of cells: connecting structure and function. Histochem Cell Biol 130:185–196
Lucic V, Rigort A, Baumeister W (2013) Cryo-electron tomography: the challenge of doing structural biology in situ. J Cell Biol 202:407–419
Ma C, Kurita D, Li N, Chen Y, Himeno H, Gao N (2017) Mechanistic insights into the alternative translation termination by ArfA and RF2. Nature 541:550–553
Mahamid J, Schampers R, Persoon H, Hyman AA, Baumeister W, Plitzko JM (2015) A focused ion beam milling and lift-out approach for site-specific preparation of frozen-hydrated lamellas from multicellular organisms. J Struct Biol 192:262–269
Mahamid J, Pfeffer S, Schaffer M, Villa E, Danev R, Cuellar LK, Forster F, Hyman AA, Plitzko JM, Baumeister W (2016) Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 351:969–972
Morgan C, Godman GC, Breitenfeld PM, Rose HM (1960) A correlative study by electron and light microscopy of the development of type 5 adenovirus. I. Electron microscopy. J Exp Med 112:373–382
Mourik MJ, Faas FG, Valentijn KM, Valentijn JA, Eikenboom JC, Koster AJ (2014) Correlative light microscopy and electron tomography to study Von Willebrand factor exocytosis from vascular endothelial cells. Methods Cell Biol 124:71–92
Oikonomou CM, Jensen GJ (2017) Cellular electron cryotomography: toward structural biology in situ. Annu Rev Biochem 86:873–896
Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, Chen SR, Yan N (2016) Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354:aah5324
Plitzko JM, Rigort A, Leis A (2009) Correlative cryo-light microscopy and cryo-electron tomography: from cellular territories to molecular landscapes. Curr Opin Biotechnol 20:83–89
Rigort A, Bauerlein FJ, Leis A, Gruska M, Hoffmann C, Laugks T, Bohm U, Eibauer M, Gnaegi H, Baumeister W, Plitzko JM (2010) Micromachining tools and correlative approaches for cellular cryo-electron tomography. J Struct Biol 172:169–179
Schorb M, Briggs JA (2014) Correlated cryo-fluorescence and cryo-electron microscopy with high spatial precision and improved sensitivity. Ultramicroscopy 143:24–32
Schorb M, Gaechter L, Avinoam O, Sieckmann F, Clarke M, Bebeacua C, Bykov YS, Sonnen AF, Lihl R, Briggs JAG (2017) New hardware and workflows for semi-automated correlative cryo-fluorescence and cryo-electron microscopy/tomography. J Struct Biol 197:83–93
Sjollema KA, Schnell U, Kuipers J, Kalicharan R, Giepmans BN (2012) Correlated light microscopy and electron microscopy. Methods Cell Biol 111:157–173
Wang H (2015) Cryo-electron microscopy for structural biology: current status and future perspectives. Sci China Life Sci 58:750–756
Wang HW, Lei J, Shi Y (2017a) Biological cryo-electron microscopy in China. Protein Sci 26:16–31
Wang S, Li S, Ji G, Huang X, Sun F (2017b) Using integrated correlative cryo-light and electron microscopy to directly observe syntaphilin-immobilized neuronal mitochondria in situ. Biophys Rep 3:8–16
Winkler H, Taylor KA (2006) Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy 106:240–254
Wolff G, Hagen C, Grunewald K, Kaufmann R (2016) Towards correlative super-resolution fluorescence and electron cryo-microscopy. Biol Cell 108:245–258
Zhang P (2013) Correlative cryo-electron tomography and optical microscopy of cells. Curr Opin Struct Biol 23:763–770
Acknowledgements
We thank the Tsinghua University Branch of the China National Center for Protein Sciences (Beijing) for providing facility support. We thank Dr. Peng Li and Dr. Xuchao Lv for providing the test samples, and Dr. Qiang Zhou for technical suggestions. This work was supported by the National Key R&D Program of China (2016YFA0501100, 2017YFA0503500).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Xiaomin Li, Jianlin Lei, and Hong-Wei Wang declare that they have no conflict of interest.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, X., Lei, J. & Wang, HW. The application of CorrSight™ in correlative light and electron microscopy of vitrified biological specimens. Biophys Rep 4, 143–152 (2018). https://doi.org/10.1007/s41048-018-0059-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41048-018-0059-x