
Abstract
Purpose of Review
Recent findings regarding major histocompatibility complex (MHC) class II polymorphisms, sources of peptides, and regulation of expression on human pathologies are described with regards to impacts on human health.
Recent Findings
Studies of MHC class II polymorphisms associated with disease indicate that beyond the selection of loaded peptide, sequence variations affect the ability of certain MHC class II to associate with chaperones that assist with peptide editing or to assume conformations needed for function. That exogenous and endogenous sources of protein are both sampled by MHC II is revealing an increasing level of overlap between classical MHC classes I and II pathways of peptide acquisition. Regulation of MHC II expression, controlled by CIITA, is also emerging as a previously underappreciated determinant of immune homeostasis.
Summary
The concept of MHC class II antigen processing and presentation to CD4 T cells has developed over the past 40 years. As research on MHC class II continues, we are beginning to discern relationships between allelic polymorphisms, peptide selection, and regulation of expression on immune health and disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Major histocompatibility complex (MHC) class II molecules present immunogenic peptides to αβ TCR bearing CD4+ T cells, an essential process for both cellular and antibody-mediated immunity. Protection against infection, vaccination, immune detection of nascent tumors, transplant rejection, constraints on autoantibody production, and even the development of these T cells all rely upon appropriate interaction with peptide:MHC II complexes. Thus, highly detailed knowledge of antigen presentation by MHC class II molecules is required to understand their function in immunity and disease, and for developing approaches to treat MHC II-associated pathologies. The general mechanisms of MHC class II antigen processing and presentation that are well-studied have been extensively reviewed [1,2,3]. Below we provide a brief overview.
MHC class II molecules (MHC II) are dimeric transmembrane proteins comprised of an alpha and a beta chain encoded in the human leukocyte antigen (HLA) locus (H-2 in mice). The most membrane distal extracellular domains of α and β form an open-ended groove capable of accommodating peptides of 13–25 amino acids in length [4]. Three different MHC isotypes are encoded in the human genome, HLA-DP, HLA-DQ, and HLA-DR (A and E in mice). Thus, most human cells expressing MHC II will commonly have two unique DP, DQ, and DR isoforms; although, some individuals have additional DR alleles. In addition, MHC II heterodimers formed by the combination of the alpha chain with both the cis and trans corresponding beta chains add to the diversity of MHC II molecules. Variations in expression and translation of DR, DP, and DQ alleles lead to unequal frequencies of MHC II expression with DR being the most abundant. Further, highly polymorphic peptide binding domain sequences within each isotype contribute to tens of thousands of allelic variations [5]. While many alleles are associated with various diseases, how allelic differences in structure or function contribute to these diseases remains unclear. Constitutive and inducible expression of MHC II genes (and most MHC II-related chaperone genes, see below) are transcriptionally regulated by a master regulator, Class II Transactivator (CIITA) [6]. CIITA itself is regulated at both the transcriptional and post-translational levels, although this is incompletely understood. While CIITA is required to regulate constitutive, cell-specific and inducible MHC II expression and contributes to T cell development and appropriate immunity, how coordinate regulation of MHC II and related genes by CIITA contributes to health and disease is likely complex and further research is needed.
So-called “professional” antigen presenting cells (APC) constitutively expressing MHC II are primarily responsible for antigen presentation to CD4+ T cells. However, other cells are capable of antigen presentation following induction of MHC II expression. Dendritic cells (DC), macrophages, and B cells are the main types of professional APC. In DC and macrophages, macropinocytosis or phagocytosis facilitates largely non-specific uptake of proteins from the extracellular millieu. In contrast, B cells primarily acquire specific extracellular proteins via antigen-specific BCR-mediated endocytosis. Once internalized, protein antigens progress through various acidic endocytic compartments containing proteases, including cathepsins, which generate peptide fragments for loading onto MHC class II. During synthesis in the ER, the chaperone invariant chain (Ii) occupies the MHC II peptide-binding groove and helps mediate endosomal trafficking [7]. In the late endosomal “MHC II compartment” (MIIC), Ii is degraded, and HLA-DM mediates exchange of the Ii CLIP (Class II-associated Ii peptide) peptide fragment present in the MHC II peptide-binding groove for other available peptides [8,9,10]. Any peptide in the MIIC is theoretically available for binding to MHC II, but chaperones HLA-DM and HLA-DO cooperate to exchange MHC II-bound peptides until a “best fit” peptide is loaded. As a result, an array of cell-surface peptide-MHC II complexes (pMHC II) are available for interaction with CD4+ T cells.
This view of MHC II antigen processing and presentation has been highly informative and shaped our present understanding of CD4 T cell-mediated immunity and immunopathology. While perhaps reflecting a prevailing mechanism for pMHC II formation, recent work is revealing novel aspects of antigen processing and presentation with unexpected complexity and highlighting other antigen processing pathways. These include polymorphisms within and outside the peptide-binding groove that contributes to the selection of peptide [11, 12], MHC II presentation of antigenic peptides derived from endogenous sources [13, 14], and the importance of regulated MHC II expression [15,16,17].
Impact of MHC Class II Polymorphisms on Peptide Selection and Immunopathology
HLA-D allele associations with susceptibility to certain infectious agents, autoimmune diseases, and immune-evasion by tumor cells are well-known [18,19,20,21,22,23]. The HLA-DP, HLA-DR, and HLA-DQ loci each contain genes coding for an alpha (DPA1, DRA1, or DQA1) and beta chain (DPB1, DRB1, or DQB1) protein. The combination of alleles on a given chromosome is called a haplotype, and since HLA genes are co-expressed, each individual expresses both a maternal and a paternal haplotype. Further HLA genes are highly polymorphic. For example, DRA1 has seven known alleles and DRB1 has over 2000 (IPD-IMGT/HLA Database, 3/2017), making for at least 14,000 potential haplotypes for HLA-DR alone. While some alleles have been sequenced completely, for others only the distal regions involved in contact with the peptide and TCR have been sequenced.
Polymorphisms and MHC Class II Structure Impacting Peptide Loading
The MHC II peptide binding groove is comprised of pockets accommodating specific amino acid residues and a network of potential hydrogen-bond formation sites that determine peptide binding specificity. The distinct dominant peptides bound by individual MHC II molecules, and affinity for these peptides is determined by polymorphisms in the binding groove. Further, while the primary alpha and beta chain sequence comprising the MHC class II binding groove is important, conformational dynamics of the molecule also impact affinity, e.g., reducing the retention of lower affinity peptides [4, 24].
In the endosome, prior to peptide loading, CLIP occupies the MHC II peptide-binding groove [25]. HLA-DM (DM) catalyzes the exchange of CLIP for antigenic peptide. Although structurally similar to other MHC class II molecules, DM does not bind peptide. Instead, DM induces conformational changes in MHC II allowing for CLIP dissociation and acquisition of higher affinity peptides. While low affinity peptides maintain a flexible conformation when in association with DM, binding of sufficiently high affinity peptide leads to greater conformational rigidity disfavoring DM-mediated exchange [12, 26]. Hence, any sequence variations affecting association with DM, either in MHC II itself or DM, is likely to impact peptide loading. Not surprisingly, DM polymorphism is less extensive (7 alpha chain and 11 beta chain variants, IPD-IMGT/HLA Database, 3/2017). However, DMA*0103, which differs at G155 and R184 relative to the more common DMA*0101 allele, exhibits dysfunctional peptide loading [27, 28]. Genetic analyses to date have failed to demonstrate DM polymorphism associations with autoimmune diseases such as rheumatoid arthritis and Type I diabetes, but the impact of DM variants likely depends also upon the combination of MHC II alleles expressed.
Polymorphisms and MHC Class II Structure Impacting Immunological Processes
While MHC II disease association studies have almost exclusively focused on peptide-binding region polymorphisms, other regions of the molecule have important immunological functions that may be relevant for disease. For instance, the transmembrane region of both alpha and beta chains contain GXXXG dimerization motif sequences important for determining the conformation of the peptide-binding/TCR contact region [29]. Duplicate GXXXG motifs in the alpha chain transmembrane region allow alternative pairing with the single motif in the beta chain, driving one of two possible conformations (M1 or M2) which likely form in human as well as mouse MHC II [11]. M1-paired MHC II are partitioned into lipid rafts [30], associate with B cell receptor signaling molecule CD79 [31], and may represent the dominant immunologically functional conformation [30].
Allelic Variations and Susceptibility to Disease
In principle, allelic variations in the peptide-binding groove that exclude critical pathogen-derived peptides might result in susceptibility to those pathogens by preventing CD4 T cell recognition and initiation of adaptive immune responses. In a Greek population exposed to or infected with West Nile Virus, DQA1*01:02 was associated with increased susceptibility to infection, and DRB1*1602 was only present in the infected population [32]. In contrast, two alleles, DQA1*0101 and DRB1*1102, were associated with complete resistance and resistance to neuro-invasion, respectively. Vaccine development efforts might also be similarly impacted by allelic differences as immunogenic peptide binding is not equivalent across alleles.
Two alleles highly associated with autoimmune idiopathic membranous nephropathy (iMN) and three amino acids within these alleles that facilitate incorporation of the dominant epitope recognized by autoantibodies in iMN patients have also been described [33]. This group further showed that the iMN-associated allele, DRB1*1501, was also associated with Goodpasture’s disease, and that two specific amino acids were critical for target-peptide binding [34]. That specific MHC II amino acid residues are associated with disease onset reinforces the concept that MHC class II structure facilitates binding of a particular peptide, which may be either protective or pathological.
While allelic variations associated with autoimmune disease can be correlated with the ability of the peptide-binding groove to accommodate endogenous antigens, some of these variations may result in poor editing by DM and thus an inability to select high affinity peptides to the MHC II peptide-binding groove. Increased stability of the DQ2 and DQ8 alleles in the beta chain alpha-helix involved in DM binding reduces peptide exchange and is associated with type 1 diabetes [24, 35]. Reduced DM binding and thus reduced peptide exchange increases the likelihood that an initial, possibly a low-affinity, endogenous peptide remains bound. In contrast, DQ1 and DQ6 are correlated with protection against type 1 diabetes due to a less stable beta chain G84-T90 region and increased DM sensitivity [24].
Sources of Peptides and Mechanisms of Peptide Generation
Sources of protein antigens and the processes generating peptides for loading onto MHC class II from those proteins are critical determinants of subsequent CD4 T cell activation and are thus important factors for understanding the impact of MHC class II presentation on health and disease. The classical source of peptides for MHC class II presentation is the extracellular environment. Phagocytosis, macropinocytosis, or receptor-mediated uptake are the main mechanisms for cellular acquisition of exogenous protein. However, peptides from endogenous (i.e., intracellular) sources have been detected on MHC class II or shown to activate CD4 T cells specific for endogenous antigen [36, 37]. These data implicate mechanisms by which intracellular peptides generated from cytosolic proteins are acquired and loaded onto endosomal MHC II. Mechanisms known to be involved in the processing of intracellular/endogenous antigens include transport of proteins to endosomal compartments by autophagy, active transport of proteasome-generated peptides to the ER, and cell surface recycling of membrane-bound receptors and associated ligands.
MHC class II presentation of endogenous peptides is expected to result in CD4 T cell tolerance. However, it can also promote autoimmunity by activating self-reactive T cells [38,39,40,41] and is an important feature of anti-viral and anti-tumor immunity [42, 43]. Appreciating more fully the sources of presented peptide and how they are generated will expand our understanding of the immunobiology of CD4 T cell selection in the thymus and responses in the periphery during infection or hypersensitivity [44]. Further, such information will likely help explain MHC II associations with autoimmune disease and improve approaches to design peptide-based therapies and vaccines.
Peptidome Analysis
The collective profile of peptides displayed on MHC class II is called the peptidome. Steps involved in analyzing the peptidome include selection of source material such as cultured cells or harvested tissues, extraction of intact MHC complexes (most commonly using monoclonal antibodies), acid-elution of peptides, separation of peptides from larger MHC II component using low molecular weight-cutoff filtration columns, resolution of peptides by chromatography, detection by mass spectrometry, and peptide identification by sequence alignment analysis [45, 46]. Improvements in biophysical methods and computational resources facilitating analysis of the peptidome have contributed to the identification of increasing numbers of MHC II-bound peptides. The potential for such analyses to define peptides critical for particular immune responses and provide therapeutic insight has yet to be fully realized owing in part to limitations in predicting the binding affinity of recovered peptides [45, 47], a lack of monoclonal antibodies recognizing the full spectrum of MHC II alleles [45], and others arising from factors such as the complexity between individuals [48]. Nevertheless, peptidome analysis has already provided new insights into the sources of the peptides found complexed with MHC class II molecules.
Sources of Peptides
The longstanding, classical understanding of MHC II antigen processing maintains that the primary source of peptides derives from exogenous proteins that are transported to degradative endosomal compartments following membrane engulfment of extracellular material [1, 2]. However, the first peptidome analysis, which used the murine B cell lymphoma cell line (LB27.4), revealed that some MHC II-displayed peptides originated from the cell itself, including peptides derived from Ii and the I-E alpha chain [36, 49]. It was proposed at the time that presentation of endogenous self-peptides might be relevant for selection of T cells during development. In fact, we now know that in the thymus, peptides generated from endogenous/intracellular AIRE-induced tissue-specific proteins and presented by MHC II are important for negative selection of autoreactive T cells [50].
Endogenous cytoplasmic antigens released into the extracellular milieu (e.g., following cell or tissue damage) can be acquired from that environment in the same fashion as extracellular proteins regardless of origin and routed to the endosomal compartment for processing. In fact, peptidome analysis of human lymph under healthy physiological conditions revealed self peptides that were of intra- and extracellular (matrix protein peptides, for example) origins [51]. By analyzing the predicted proteolytic cleavage sites of these lymph-derived peptides, proteases in addition to endosomal cathepsins, including MMPs and caspases, were suggested to be involved in peptide generation [51]. A comparison of peptides extracted from lymph and those eluted from MHC class II on HLA-DR1 of DC showed much overlap indicating that proteins cleaved in the lymph were being presented on MHC class II [51]. Proposed mechanisms for loading these extracellular peptides derived from intracellular self-proteins include exchange of peptides at the cell surface and/or exchange within recycling/sorting endosomes [52,53,54,55]. In either mechanism, the previously loaded peptide must be displaced in favor of the acquired peptide. Assuming that the surface and recycling MHC class II molecules involved had already undergone successful exchange for high-affinity peptide, how this process accomplishes such exchange remains unclear.
Identification of viral antigen-derived peptides on MHC class II has also provided insight into mechanisms that facilitate presentation of cytosolic [56]. Peptides generated in the cell cytoplasm can be targeted to MHC II through autophagy-mediated transport into the late endosomal compartment and has recently been reviewed [13, 57, 58]. In addition to autophagy, proteosome-dependent mechanisms like those use by MHC class I are an additional means of processing endogenous peptides for presentation by MHC II [14, 59]. In order for cytoplasmic peptides generated via proteasomes to be presented on MHC class II, the peptide must likely encounter MHC II-lacking bound peptide. While MHC II is in the ER, the peptide-binding groove is generally thought to be occupied by CLIP until transport to endosomes where CLIP is exchanged for peptide by DM. If the peptide-binding groove is unoccupied while MHC II resides in the ER, proteasome-elaborated peptides entering the ER via the transporter association with [MHC class I] antigen processing (TAP) could be bound by MHC II. In fact, alleles of HLA-DP bearing the beta chain polymorphism 84Gly, although still transported normally by Ii, fail to bind CLIP, are empty while located in the ER, and arrive at the cell surface complexed with endogenously derived peptide [60]. In another study, Becker et al. demonstrated that certain MHC II peptides presented by CD40-activated B cells were processed by proteasomes following delivery of pinocytosed antigen [61]. Because other peptides from the same antigen can be presented by MHC I, it was suggested that strict separation of endogenous and exogenous presentation by MHC I and MHC II, respectively, might be an oversimplification. Better understanding of how endogenous antigens are processed and presented might allow for optimization of vaccines and tumor immunotherapies. For example, targeting the HIV gag protein to autophagosomes by fusion to LC3 led to enhanced T cell activation which might represent a useful approach to promoting effective anti-HIV immunity [62].
B cells are somewhat unique as APCs as they express a clonally restricted antigen receptor, the BCR. While B cells can process and present high concentration environmental (e.g., self) antigens internalized by fluid-phase endocytosis, BCR-mediated processing of cognate antigen is of critical importance. Recently, it was shown that internalized antigen-BCR (Ag-BCR) complexes physically associated with intracellular MHC class II molecules in a putative MHC class II peptide-loading complex (PLC) [31]. Interestingly, the class II molecules that associate with the Ag-BCR complexes are a unique conformational subset (i.e., M1 paired class II, see above), and peptide derived from the processing of BCR-bound antigen are selectively presented in the context of these M1-paired class II molecules. In contrast, peptide from antigen acquired by fluid-phase endocytosis is loaded onto both M1 and M2 paired class II [63]. This finding is consistent with the past work of Mitchell and colleagues who found that peptides derived from BCR-bound cognate antigen compete for binding to a pool of MHC class II molecules that cannot be accessed by peptides generated from antigens internalized by fluid-phase uptake [64]. The findings could also explain the differential B cell response to engagement of peptide-class II complexes formed via BCR vs. fluid-phase antigen processing [65]. Thus, similar to MHC class I molecules, where peptide loading occurs in a complex including “accessory” molecules such as TAP and tapasin, MHC class II peptide loading may occur in a complex including HLA-DM, a source of peptide, and other yet-to-be identified “accessory” molecules.
Regulation of MHC Class II Expression
Without expression of MHC class II molecules, the source of antigenic peptide and structure of the presenting complex are irrelevant. Much of our understanding of MHC class II expression follows from the study of human MHC class II deficiency. A congenic lack of MHC class II expression results in the general failure of CD4+ T cell development and leads to a severe combined immunodeficiency (bare lymphocyte syndrome, Type II (BLS)) generally causing death within the first year of life [66]. Failed transcription of MHC class II genes accounts for all instances of Type II BLS, and the transcription factors essential for the constitutive and inducible expression of MHC class II and MHC class II-related proteins are known. A global regulator of MHC class II gene transcription, the class II transactivator (CIITA), controls both constitutive expression in professional antigen-presenting cells and inducible expression in most other cells and tissues. CIITA directs the transcription of not only MHC class II genes, but the chaperones Ii and DM as well as Rab4b, a small GTPase protein involved in endosomal recycling [67, 68]. Regulation by CIITA is thus a central mechanism impacting the selection and presentation of antigen peptide. Further, altered MHC class II expression is a feature common in infectious disease, autoimmunity, and cancer.
Human CIITA has four distinct promoters and mouse C2ta has three, which control the expression of three different CIITA isoforms [69, 70]. These distinct promoters facilitate both constitutive and inducible expression and drive cell-type specific expression patterns. Antigen-presenting cells express multiple isoforms of CIITA, but the pattern differs by cell type and activation status. Promoter I-driven CIITA isoform I is most abundant in dendritic cells and macrophages and contains a caspase-recruitment domain (CARD) that increases MHC II transcription [71]. Isoform III (promoter III) is constitutively expressed in B cells and pDCs, and isoform IV (promoter IV) is induced by IFNγ in both hematopoietic and non-hematopoietic cells. CIITA isoforms III and IV lack the CARD domain, but otherwise differ only in that isoform IV lacks the first 24 residues present in isoform III [69]. However, how the isoforms differ functionally, why some cells express multiple isoforms, how differential regulation of isoform expression impacts adaptive immunity, and even why such a complex system is beneficial remain largely unaddressed.
While HLA polymorphisms have long been the subject of disease association studies, genetic variations in CIITA have received less attention. Mouse variants in C2ta promoter I that increase MHC II expression in myeloid cells, but not B cells, from spleen and blood, have recently been described [17]. IFN-gamma-treated bone marrow-derived macrophages from mice carrying this variant also expressed more MHC II. Increased MHC class II expression correlated with greater T cell activation, but disease development in experimental models of autoimmune disease (collagen-induced arthritis and EAE) was unaffected, suggesting that while this C2ta polymorphism alters antigen presenting cell MHC II expression, it may not greatly influence autoimmunity [17]. In contrast, in a mouse model of experimental autoimmune myocarditis (EAM), IFN-gamma from autoreactive T cells induced the promoter IV form of CIITA in endothelial cells [72]. Induced endothelial MHC II in turn presented cardiac antigens to effector T cells resulting in a cyclic and progressive cardiomyopathy. Neutralization of IFN-γ or deletion of C2ta promoter IV prevented MHC II expression on non-hematopoietic cells and reduced indicators of cardiomyopathy [72]. Increased expression of MHC class II on endothelial cells has been demonstrated for other autoimmune diseases including RA and SLE [73]. Although not interfering with CIITA function, specific deletion of MHC II expression in cDC in mice revealed a role for these APCs in maintaining homeostasis of the gut microbiota [11]. These cDCs were still responsive to intestinal microbes, but defective T cell help for germinal center B cell class switching to IgA accounted for increased chronic intestinal inflammation that was alleviated with antibiotics or housing under germ-free conditions [11].
Dysregulated expression of MHC II may be harmful in autoimmune conditions, but MHC II expression on tumor cells appears beneficial in some instances. In a study of triple-negative breast cancer (a particularly aggressive tumor and not responsive to estrogen receptor and HER2/neu targeted therapy), increased tumor cell expression of MHC II and related genes including CIITA, CD74 (Ii), and HLA was associated with progression-free survival (a good prognosis) [74]. Tumor-infiltrating T and B cells were also increased in tumors associated with good prognosis indicating that presentation of tumor antigens on MHC class II leads to antitumor immune responses. The TS/A mouse mammary adenocarcinoma model (which resembles triple negative breast cancer) recapitulates favorable anti-tumor immunity when MHC II is expressed [75, 76]. In a study on diffuse large B cell lymphoma, good prognosis was also correlated with MHC II gene expression, specifically HLA-DR and CD74 [77]. Here, expression of the transcription factor FOXP1 was correlated with decreased expression of MHC class II genes, possibly by binding promoters of CIITA responsive genes. Therapies-stimulating expression of CIITA, including the use of histone deacetylase inhibitors to restore MHC II expression, are being investigated for use in combatting tumors [78, 79]
While it is generally held that only a few hundred MHC II:peptide complexes are necessary for successful antigen presentation, the effects of reduced MHC II expression are not well-studied. One group generated a mouse strain with an eightfold reduction in global class II expression [15]. While a complete absence of MHC II results in deficient development of CD4+ T cells, an eightfold reduction had no appreciable effect on CD4+ or Treg development in the thymus. In addition, normal functions of CD4 and Treg were observed. However, liver-specific autoreactive T cells were increased [15], suggesting that decreased MHC II expression, perhaps in conjunction with AIRE-induced expression of a restricted set of tissue-specific antigens, resulted in diminished negative selection of these tissue-specific autoreactive T cells. Therefore, while maximal expression is not a requirement for development, reduced levels impact the ability to present sufficient auto antigens to effectively remove maturing T cells specific to peripheral antigens. As MHC II expression is globally regulated by CIITA, attention to CIITA variants with reduced transactivation function, conditions that reduce the expression of function of CIITA, and the detailed mechanisms of CIITA function are important, but understudied areas of investigation.
Conclusion
Although much is known about MHC II antigen processing and presentation, how the intricacies of this system result in disease is not understood. Polymorphic variations in MHC class II structure affect peptide loading, but whether or not specific bound peptides are ultimately and solely responsible for various pathologies, such as autoimmune disease, tumor evasion, and consequences of infectious disease, is unclear. Structures outside the peptide-binding groove which drive subtle structural features marking potentially functionally distinct subsets of MHC II proteins are also important. Increasing evidence indicates overlap between the classical MHC I and MHC II presentation pathways consistent with MHC II loading with both endogenous and exogenous peptide and with implications for directing immune responses to prevent or treat disease. Finally, MHC II expression by specific cell types and quantities of surface MHC II, controlled largely by the transcription factor CIITA, may be responsible for distinct immunologic functions. Further discoveries in these areas will likely transform our present understanding of how MHC II influences both health and disease.
References
Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–73. doi:10.1146/annurev-immunol-032712-095910.
Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. 2015;15(4):203–16. doi:10.1038/nri3818.
Unanue ER, Turk V, Neefjes J. Variations in MHC class II antigen processing and presentation in health and disease. Annu Rev Immunol. 2016;34:265–97. doi:10.1146/annurev-immunol-041015-055420.
Wieczorek M, Abualrous ET, Sticht J, Alvaro-Benito M, Stolzenberg S, Noe F, et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. 2017;8:292. doi:10.3389/fimmu.2017.00292.
Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet. 2009;54(1):15–39. doi:10.1038/jhg.2008.5.
Devaiah BN, Singer DS. CIITA and its dual roles in MHC gene transcription. Front Immunol. 2013;4:476. doi:10.3389/fimmu.2013.00476.
Roche PA, Marks MS, Cresswell P. Formation of a nine-subunit complex by HLA class II glycoproteins and the invariant chain. Nature. 1991;354(6352):392–4. doi:10.1038/354392a0.
Mosyak L, Zaller DM, Wiley DC. The structure of HLA-DM, the peptide exchange catalyst that loads antigen onto class II MHC molecules during antigen presentation. Immunity. 1998;9(3):377–83.
Roche PA, Cresswell P. Proteolysis of the class II-associated invariant chain generates a peptide binding site in intracellular HLA-DR molecules. Proc Natl Acad Sci U S A. 1991;88(8):3150–4.
Romagnoli P, Germain RN. The CLIP region of invariant chain plays a critical role in regulating major histocompatibility complex class II folding, transport, and peptide occupancy. J Exp Med. 1994;180(3):1107–13.
Drake LA, Drake JR. A triad of molecular regions contribute to the formation of two distinct MHC class II conformers. Mol Immunol. 2016;74:59–70. doi:10.1016/j.molimm.2016.04.010.
Ferrante A, Templeton M, Hoffman M, Castellini MJ. The thermodynamic mechanism of peptide-MHC class II complex formation is a determinant of susceptibility to HLA-DM. J Immunol. 2015;195(3):1251–61. doi:10.4049/jimmunol.1402367.
Duraes FV, Niven J, Dubrot J, Hugues S, Gannage M. Macroautophagy in endogenous processing of self- and pathogen-derived antigens for MHC class II presentation. Front Immunol. 2015;6:459. doi:10.3389/fimmu.2015.00459.
Leung CS. Endogenous antigen presentation of MHC class II epitopes through non-autophagic pathways. Front Immunol. 2015;6:464. doi:10.3389/fimmu.2015.00464.
Chen YT, Su YC, Chang ML, Tsai PF, Kung JT. Low-level MHC class II expression leads to suboptimal Th cell response, increased autoaggression, and heightened cytokine inducibility. J Immunol. 2017;198(5):1928–43. doi:10.4049/jimmunol.1600967.
Loschko J, Schreiber HA, Rieke GJ, Esterhazy D, Meredith MM, Pedicord VA, et al. Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. J Exp Med. 2016;213(4):517–34. doi:10.1084/jem.20160062.
Yau AC, Piehl F, Olsson T, Holmdahl R. Effects of C2ta genetic polymorphisms on MHC class II expression and autoimmune diseases. Immunology. 2017;150(4):408–17. doi:10.1111/imm.12692.
Deitiker P, Atassi MZ. MHC genes linked to autoimmune disease. Crit Rev Immunol. 2015;35(3):203–51.
Lee HJ, Li CW, Hammerstad SS, Stefan M, Tomer Y. Immunogenetics of autoimmune thyroid diseases: a comprehensive review. J Autoimmun. 2015;64:82–90. doi:10.1016/j.jaut.2015.07.009.
Lenz TL, Deutsch AJ, Han B, Hu X, Okada Y, Eyre S, et al. Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat Genet. 2015;47(9):1085–90. doi:10.1038/ng.3379.
Mosaad YM. Clinical role of human leukocyte antigen in health and disease. Scand J Immunol. 2015;82(4):283–306. doi:10.1111/sji.12329.
Noble JA. Immunogenetics of type 1 diabetes: a comprehensive review. J Autoimmun. 2015;64:101–12. doi:10.1016/j.jaut.2015.07.014.
Tsai S, Santamaria P. MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front Immunol. 2013;4:321. doi:10.3389/fimmu.2013.00321.
Wieczorek M, Sticht J, Stolzenberg S, Gunther S, Wehmeyer C, El Habre Z, et al. MHC class II complexes sample intermediate states along the peptide exchange pathway. Nat Commun. 2016;7:13224. doi:10.1038/ncomms13224.
Roche PA, Cresswell P. Invariant chain association with HLA-DR molecules inhibits immunogenic peptide binding. Nature. 1990;345(6276):615–8. doi:10.1038/345615a0.
Sant AJ, Chaves FA, Jenks SA, Richards KA, Menges P, Weaver JM, et al. The relationship between immunodominance, DM editing, and the kinetic stability of MHC class II:peptide complexes. Immunol Rev. 2005;207:261–78. doi:10.1111/j.0105-2896.2005.00307.x.
Alvaro-Benito M, Morrison E, Wieczorek M, Sticht J, Freund C. Human leukocyte Antigen-DM polymorphisms in autoimmune diseases. Open Biol. 2016;6(8). doi:10.1098/rsob.160165.
Alvaro-Benito M, Wieczorek M, Sticht J, Kipar C, Freund C. HLA-DMA polymorphisms differentially affect MHC class II peptide loading. J Immunol. 2015;194(2):803–16. doi:10.4049/jimmunol.1401389.
King G, Dixon AM. Evidence for role of transmembrane helix-helix interactions in the assembly of the class II major histocompatibility complex. Mol BioSyst. 2010;6(9):1650–61. doi:10.1039/c002241a.
Busman-Sahay K, Sargent E, Harton JA, Drake JR. The Ia.2 epitope defines a subset of lipid raft-resident MHC class II molecules crucial to effective antigen presentation. J Immunol. 2011;186(12):6710–7. doi:10.4049/jimmunol.1100336.
Barroso M, Tucker H, Drake L, Nichol K, Drake JR. Antigen-B cell receptor complexes associate with intracellular major histocompatibility complex (MHC) class II molecules. J Biol Chem. 2015;290(45):27101–12. doi:10.1074/jbc.M115.649582.
Sarri CA, Markantoni M, Stamatis C, Papa A, Tsakris A, Pervanidou D, et al. Genetic contribution of MHC class II genes in susceptibility to West Nile virus infection. PLoS One. 2016;11(11):e0165952. doi:10.1371/journal.pone.0165952.
Cui Z, Xie LJ, Chen FJ, Pei ZY, Zhang LJ, Qu Z, et al. MHC class II risk alleles and amino acid residues in idiopathic membranous nephropathy. J Am Soc Nephrol. 2016; doi:10.1681/ASN.2016020114.
Xie LJ, Cui Z, Chen FJ, Pei ZY, Hu SY, Gu QH, et al. The susceptible HLA class II alleles and their presenting epitope(s) in Goodpasture’s disease. Immunology. 2017; doi:10.1111/imm.12736.
Zhou Z, Reyes-Vargas E, Escobar H, Rudd B, Rockwood AL, Delgado JC, et al. Type 1 diabetes associated HLA-DQ2 and DQ8 molecules are relatively resistant to HLA-DM mediated release of invariant chain-derived CLIP peptides. Eur J Immunol. 2016;46(4):834–45. doi:10.1002/eji.201545942.
Rudensky AY, Preston-Hurlburt P, Hong SC, Barlow A, Janeway CA Jr. Sequence analysis of peptides bound to MHC class II molecules. Nature. 1991;353:622–627.
Sant AJ. Endogenous antigen presentation by MHC class II molecules. Immunol Res. 1994;13(4):253–67.
Palma ML, Duangkhae P, Douradinha B, Viana IFT, Rigato PO, Dhalia R, et al. Development of potent class II transactivator gene delivery systems capable of inducing de novo MHC II expression in human cells, in vitro and ex vivo. Gene Ther. 2017; doi:10.1038/gt.2017.25.
Parnell GP, Booth DR. The multiple sclerosis (MS) genetic risk factors indicate both acquired and innate immune cell subsets contribute to MS pathogenesis and identify novel therapeutic opportunities. Front Immunol. 2017;8:425. doi:10.3389/fimmu.2017.00425.
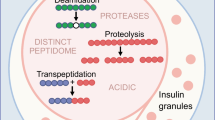
Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11(4):350–4. doi:10.1038/ni.1850.
Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med. 2011;208(12):2375–83. doi:10.1084/jem.20111502.
Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54(8):721–8. doi:10.1007/s00262-004-0653-2.
Miller MA, Ganesan AP, Luckashenak N, Mendonca M, Eisenlohr LC. Endogenous antigen processing drives the primary CD4+ T cell response to influenza. Nat Med. 2015;21(10):1216–22. doi:10.1038/nm.3958.
Collado JA, Guitart C, Ciudad MT, Alvarez I, Jaraquemada D. The repertoires of peptides presented by MHC-II in the thymus and in peripheral tissue: a clue for autoimmunity? Front Immunol. 2013;4:442. doi:10.3389/fimmu.2013.00442.
Schumacher FR, Delamarre L, Jhunjhunwala S, Modrusan Z, Phung QT, Elias JE et al. Building proteomic tool boxes to monitor MHC class I and class II peptides. Proteomics. 2017;17(1–2). doi:10.1002/pmic.201600061.
Sofron A, Ritz D, Neri D, Fugmann T. High-resolution analysis of the murine MHC class II immunopeptidome. Eur J Immunol. 2016;46(2):319–28. doi:10.1002/eji.201545930.
Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4(4):e1000048. doi:10.1371/journal.pcbi.1000048.
Fugmann T, Sofron A, Ritz D, Bootz F, Neri D. The MHC class II immunopeptidome of lymph nodes in health and in chemically induced colitis. J Immunol. 2017;198(3):1357–64. doi:10.4049/jimmunol.1601157.
Unanue ER. The secrets of the class II MHC peptidome start to be revealed. J Immunol. 2016;196(3):939–40. doi:10.4049/jimmunol.1502571.
Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298(5597):1395–401. doi:10.1126/science.1075958.
Clement CC, Becerra A, Yin L, Zolla V, Huang L, Merlin S, et al. The dendritic cell major histocompatibility complex II (MHC II) peptidome derives from a variety of processing pathways and includes peptides with a broad spectrum of HLA-DM sensitivity. J Biol Chem. 2016;291(11):5576–95. doi:10.1074/jbc.M115.655738.
Call MJ, Xing X, Cuny GD, Seth NP, Altmann DM, Fugger L, et al. In vivo enhancement of peptide display by MHC class II molecules with small molecule catalysts of peptide exchange. J Immunol. 2009;182(10):6342–52. doi:10.4049/jimmunol.0803464.
Nicholson MJ, Moradi B, Seth NP, Xing X, Cuny GD, Stein RL, et al. Small molecules that enhance the catalytic efficiency of HLA-DM. J Immunol. 2006;176(7):4208–20.
Pos W, Sethi DK, Wucherpfennig KW. Mechanisms of peptide repertoire selection by HLA-DM. Trends Immunol. 2013;34(10):495–501. doi:10.1016/j.it.2013.06.002.
Schulze MS, Wucherpfennig KW. The mechanism of HLA-DM induced peptide exchange in the MHC class II antigen presentation pathway. Curr Opin Immunol. 2012;24(1):105–11. doi:10.1016/j.coi.2011.11.004.
Veerappan Ganesan AP, Eisenlohr LC. The elucidation of non-classical MHC class II antigen processing through the study of viral antigens. Curr Opin Virol. 2017;22:71–6. doi:10.1016/j.coviro.2016.11.009.
Munz C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. 2016;37(11):755–63. doi:10.1016/j.it.2016.08.017.
Munz C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol Rev. 2016;272(1):17–27. doi:10.1111/imr.12422.
Tewari MK, Sinnathamby G, Rajagopal D, Eisenlohr LC. A cytosolic pathway for MHC class II-restricted antigen processing that is proteasome and TAP dependent. Nat Immunol. 2005;6(3):287–94. doi:10.1038/ni1171.
Yamashita Y, Anczurowski M, Nakatsugawa M, Tanaka M, Kagoya Y, Sinha A, et al. HLA-DP84Gly constitutively presents endogenous peptides generated by the class I antigen processing pathway. Nat Commun. 2017;8:15244. doi:10.1038/ncomms15244.
Becker HJ, Kondo E, Shimabukuro-Vornhagen A, Theurich S, von Bergwelt-Baildon MS. Processing and MHC class II presentation of exogenous soluble antigen involving a proteasome-dependent cytosolic pathway in CD40-activated B cells. Eur J Haematol. 2016;97(2):166–74. doi:10.1111/ejh.12699.
Coulon PG, Richetta C, Rouers A, Blanchet FP, Urrutia A, Guerbois M, et al. HIV-infected dendritic cells present endogenous MHC class II-restricted antigens to HIV-specific CD4+ T cells. J Immunol. 2016;197(2):517–32. doi:10.4049/jimmunol.1600286.
Harton J, Jin L, Hahn A, Drake J. Immunological Functions of the Membrane Proximal Region of MHC Class II Molecules. F1000Res. 2016;5. doi:10.12688/f1000research.7610.1.
Mitchell RN, Barnes KA, Grupp SA, Sanchez M, Misulovin Z, Nussenzweig MC, et al. Intracellular targeting of antigens internalized by membrane immunoglobulin in B lymphocytes. J Exp Med. 1995;181(5):1705–14.
Nashar TO, Drake JR. The pathway of antigen uptake and processing dictates MHC class II-mediated B cell survival and activation. J Immunol. 2005;174(3):1306–16.
Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol. 2001;19:331–73. doi:10.1146/annurev.immunol.19.1.331.
Harton JA, Ting JP. Class II transactivator: mastering the art of major histocompatibility complex expression. Mol Cell Biol. 2000;20(17):6185–94.
Krawczyk M, Leimgruber E, Seguin-Estevez Q, Dunand-Sauthier I, Barras E, Reith W. Expression of RAB4B, a protein governing endocytic recycling, is co-regulated with MHC class II genes. Nucleic Acids Res. 2007;35(2):595–605. doi:10.1093/nar/gkl980.
Muhlethaler-Mottet A, Otten LA, Steimle V, Mach B. Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J. 1997;16(10):2851–60. doi:10.1093/emboj/16.10.2851.
Pai RK, Askew D, Boom WH, Harding CV. Regulation of class II MHC expression in APCs: roles of types I, III, and IV class II transactivator. J Immunol. 2002;169(3):1326–33.
Nickerson K, Sisk TJ, Inohara N, Yee CS, Kennell J, Cho MC, et al. Dendritic cell-specific MHC class II transactivator contains a caspase recruitment domain that confers potent transactivation activity. J Biol Chem. 2001;276(22):19089–93. doi:10.1074/jbc.M101295200.
Thelemann C, Haller S, Blyszczuk P, Kania G, Rosa M, Eriksson U, et al. Absence of nonhematopoietic MHC class II expression protects mice from experimental autoimmune myocarditis. Eur J Immunol. 2016;46(3):656–64. doi:10.1002/eji.201545945.
Turesson C. Endothelial expression of MHC class II molecules in autoimmune disease. Curr Pharm Des. 2004;10(2):129–43.
Forero A, Li Y, Chen D, Grizzle WE, Updike KL, Merz ND, et al. Expression of the MHC class II pathway in triple-negative breast cancer tumor cells is associated with a good prognosis and infiltrating lymphocytes. Cancer Immunol Res. 2016;4(5):390–9. doi:10.1158/2326-6066.CIR-15-0243.
Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol. 2003;33(5):1183–92. doi:10.1002/eji.200323712.
Mortara L, Castellani P, Meazza R, Tosi G, De Lerma BA, Procopio FA, et al. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin Cancer Res. 2006;12(11 Pt 1):3435–43. doi:10.1158/1078-0432.CCR-06-0165.
Brown PJ, Wong KK, Felce SL, Lyne L, Spearman H, Soilleux EJ, et al. FOXP1 suppresses immune response signatures and MHC class II expression in activated B-cell-like diffuse large B-cell lymphomas. Leukemia. 2016;30(3):605–16. doi:10.1038/leu.2015.299.
Bou Nasser Eddine F, Forlani G, Lombardo L, Tedeschi A, Tosi G, Accolla RS. CIITA-driven MHC class II expressing tumor cells can efficiently prime naive CD4+ TH cells in vivo and vaccinate the host against parental MHC-II-negative tumor cells. Oncoimmunology. 2017;6(1):e1261777. doi:10.1080/2162402X.2016.1261777.
Turner TB, Meza-Perez S, Londono A, Katre A, Peabody JE, Smith HJ, et al. Epigenetic modifiers upregulate MHC II and impede ovarian cancer tumor growth. Oncotarget. 2017; doi:10.18632/oncotarget.17395.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ellen B. Duffy, James R. Drake, and Jonathan A. Harton declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Immunology and Inflammation
Rights and permissions
About this article

Cite this article
Duffy, E.B., Drake, J.R. & Harton, J.A. Evolving Insights for MHC Class II Antigen Processing and Presentation in Health and Disease. Curr Pharmacol Rep 3, 213–220 (2017). https://doi.org/10.1007/s40495-017-0097-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-017-0097-y