
Abstract
Targeted drug delivery is critical for minimizing off-target toxicity by chemotherapeutic drugs. Amphotericin-B is an effective anti-leishmaniasis agent that induces significant nephrotoxicity. Complexing amphotericin-B with liposomes (AmBisome, LAmB) reduces nephrotoxicity and improves LAmB efficacy in treating leishmaniasis in humans. However, complicated dosing regimens are required to minimize side effects. Given that Leishmania species are obligate parasites in phagocytes, selectively targeting LAmB to infected phagocytes would likely improve efficacy, decrease systemic toxicity, and simplify dosing regimens. To target LAmB to phagocytes, we developed a PADRE-derivatized-dendrimer (PDD)-LAmB nanocarrier platform by functionalizing dendrimers that complex LAmB with a peptide that targets major histocompatibility complex (MHC) class II receptors. MHC class II receptor expression increases on phagocytes during the course of leishmaniasis, further enhancing infected cells as targets for PDD-LAmB. In a murine model for Leishmania major infection, PDD-LAmB efficiently targeted infected phagocytic cells, reducing the LAmB effective dose by 80 %, with improved pharmacokinetics. Moreover, the PDD MHC class II-targeting peptide is a universal helper T-cell epitope, allowing PDD-LAmB complexes to elicit parasite-specific T-cell responses. Thus, PDD-LAmB complex is a promising candidate for further development and human clinical trials, reducing and simplifying LAmB dosing, reducing side effects, and stimulating T-cell responses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Leishmaniasis is a neglected global health concern that is endemic in 88 countries. There is currently no vaccine, and the limited therapeutic options also have significant adverse side effects [1]. The immunopathology induced by Leishmania species ranges from the cutaneous to the visceral forms, depending on the species of Leishmania and the host immune response. Leishmaniasis was previously observed mainly in farming communities and the military. However, today the transmission of this disease has spread to cities, in particular in Asia, Africa, and the Americas. Leishmaniasis has also been reported among immunosuppressed individuals, a growing concern due to chronic viral infections including HIV, organ transplantation, and associated treatment with immunosuppressive drugs. Cases of leishmaniasis have recently been reported in countries where previously it was rarely observed, including France, Italy, Spain, and Portugal.
Leishmania parasites are obligatory intracellular microbes that have the ability to survive and multiply inside the phagolysosomal environment of infected macrophages and dendritic cells. Importantly, dendritic cells and macrophages are antigen-presenting cells (APCs) that dictate the fate of the immune response to the pathogen. The cellular immune response against leishmania generated by these APCs is of particular concern. Cellular immunity is required for clearance of infected cells, given that antibodies cannot gain access into cells, and only antigen-specific T cells can identify and kill cells infected with intracellular pathogens. Indeed, APC induction dictates whether immune cells will mount adaptive and memory responses and the magnitude of such responses. T-helper cells are also critical for this response and have the ability to orchestrate innate/adaptive effector cell activity. T-helper cells influence almost every aspect of an anti-leishmania immune response. For example, they secrete necessary cytokines, induce B cells to undergo class switching to mature antibody responses, expand and maintain cluster of differentiation (CD)-8+ T-cell responses, enhance the recruitment of innate effector cells, and help increase the breadth of the immune responses [2].
Importantly, protective immunity against these obligatory intracellular parasites requires T-helper 1 (Th1 type) cellular immune responses, including interferon (IFN)-γ-secreting CD4 T cells and cytotoxic CD8 T cells [1, 3]. To blunt the generation of protective immunity, leishmania parasites use immune-avoidance mechanisms, including changes in interleukin (IL)-10 expression [4]. Furthermore, early in the infection, promastigotes of Leishmania major mainly target resident macrophages. Infection of resident macrophages has been shown to cause a cellular stress response and expression of pro-inflammatory signals. These signals promote parasite survival and replication in host macrophages [5]. However, more recently it has been shown that Th1/Th2 cells, and their relationship with IL-4 and IL-12, does not fully explain the complex leishmaniasis immunopathology; it was shown, for example, that a Th2-like response could occur in the absence of IL-4 and, under certain circumstances, IL-4 can prime for IL-12 expression [6].
Macrophage infection by leishmania is mediated by complement receptor (CR)-3, and infection by L. major was shown to inhibit IL-12 production by engaging CR3 [7]. Leishmania species thus preferentially gain access to cells that can initiate adaptive immunity against them. In both humans and mice, protection against L. major infection is correlated with antigen-specific Th1-like CD4+ T cells. Interestingly, antibody responses against leishmania also play an important role by forming complexes with the parasite that result in increased dendritic cell uptake. This uptake results in a Th1 response and expression of IL-12. Arguably, the consensus is that IL-12 is needed for protection, and its production from infected cells induces natural killer (NK) cell activation and CD4+ Th1 expansion, which produces IFN-γ. Macrophages become stimulated and make inducible nitric oxide synthase (iNOS), resulting in parasite killing. In cases where a predominant IL-4/IL-13/IL-10 response is induced, parasite replication is not controlled. CD4+ CD25+ T regulatory cells are a main producer of IL-10 and contribute to disease persistence. The challenge of generating a strong Th1 immune response has delayed production of an effective leishmania vaccine.
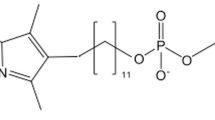
Similarly, chemotherapy of leishmaniasis is hampered by a lack of effective treatment options. Currently available drugs for leishmaniasis show poor efficacy, low bioavailability, low stability, adverse effects, and high cost. These drugs, many more than 40 years old, are in need of improvement or the development of alternative, novel approaches. One recent improvement has been the use of liposomal amphotericin B (LAmB), a drug for the prophylaxis and treatment of fungal infections, the treatment of visceral leishmaniasis, and, more recently, the treatment of cutaneous leishmaniasis. Nevertheless, even LAmB is associated with toxicity, and the LAmB package insert cites nephrotoxicity and neurologic toxicities. LAmB also requires administration via infusion in clinics, and must be followed by intravenous administration of 2 liters of saline to reduce its nephrotoxicity.
Novel Approaches
Today’s drug delivery methods are very inefficient, with, in some cases, as little as 1 % of the drug reaching its intended organ/cells, resulting in increased toxicities, complicated dosage, higher costs and liabilities, and the attrition of approximately one-third of drug candidates [8]. The success rate of first-in-human to registration is indeed only 11 %, mainly due to low efficacy and toxicity [8]. Combinatorial chemotherapy and immunotherapy have been proposed as novel means to enhance anti-leishmania drug efficacy. An effective Th1 immune response plays a crucial role in the control of leishmaniasis, causing the induction of Th1 cytokines that allow the recruitment of dendritic cells and T-effector cells. Thus, enhancing a Th1 immune response should be additive or synergistic with chemotherapy. For example, the use of amphotericin B (Amp B) together with IL-12, anti-CD40 antibodies, or anti-IL-10 antibodies was able to significantly increase the parasite-killing effect of the drug [9].
Chemotherapy of leishmania infections started with antimonial compounds for both the cutaneous (CL) and visceral (VL) forms. Later, other drugs such as pentamidine, ketoconazole, itraconazole, and fluconazole were used with varying results. Miltefosine was the first active oral drug but is active for VL. Amp B formulations (Fungizone®, Abelcet ®, and AmBisome®) are available but need to be injected. Paromomycin is used as a topical agent for CL and by intramuscular injections for VL [10, 11]. New formulations of nanoparticles and Amp B have been used with varying results in hamsters infected with VL. These formulations include phosphoserine-coated gelatin nanoparticles coupled with Amp B, PLGA-Peg encapsulated Amp B nanoparticles and Amp B-encapsulated chitosan nanocapsules, which also worked as an immunoadjuvant in up-regulating tumor necrosis factor, IL-12, and iNOS [12]. Combination therapy for VL in humans has shown good activity for these combinations: AmBisome + miltefosine, paromomycin + miltefosine and antimonials + paromomycin [13]. However, targeting the cells that serve as the reservoir of leishmania parasites has rarely been explored.
Given that only a fraction of LAmB reaches the intended target cells following systemic administration, we postulated that selective delivery of LAmB to phagocytes would lower the off-target effects of Amp B and reduce its toxicity. Furthermore, the encapsulating of the drugs in nanocarriers limits interaction with proteins, hormones, and other components in the host, further limiting undesired drug reactions. A LAmB nanocarrier targeting APCs would be ideally suited to reduce the drug dosage and regimen requirements during leishmaniasis treatment. We recently reported that LAmB conjugated to a nanocarrier results in shortening of cutaneous leishmaniasis in a mouse model, while also enhancing L. major-specific T-cell immune responses in the infected host.
PDD-LAmB
We have created a nanocarrier, PADRE-derivatized-dendrimer (PDD), for targeting of LAmB to macrophages and dendritic cells. The net positively charged PDD is able to complex with negatively charged LAmB (Fig. 1). Since PDD has an intrinsic targeting moiety allowing it to home to macrophages and dendritic cells, it reduces off-target effects of LAmB. In addition, PDD is a Th1 inducer, and therefore it can both target macrophages/dendritic cells and enhance host T-cell responses against leishmanial antigens expressed by infected macrophages [14••]. Harnessing host immunity combined with chemotherapy should further reduce the dose of LAmB required.

PDDs, APC targeting nanocarrier, and PDD-drug. Drug-carrying APC-targeting platform elicits TH1 responses in the presence of antigen. Two formulations have been created: ‘1st formulation’ uses non-covalent interactions (including electrostatic or hydrophobic interactions) for the engraftment of the drugs into PDD; ‘2nd formulation’ is composed of the drugs that are encapsulated into negatively charged liposome, which is loaded with positively charged PDD. A drug-carrying nanocarrier platform that targets cells expressing MHC class II. Macrophages express MHC class II and are the reservoir for Leishmania parasites. PDDs platform for homing drugs to cells expressing MHC class II (APC). Peptides that bind with high affinity to APCs (macrophages, monocytes, Langerhans cells, and B cells) are covalently linked to the surface-modified dendrimers. The platform is positively charged and makes a stable complex with negatively charged drugs, e.g. LAmB. Alternatively, drugs may be complexed with PDD via hydrophobic interactions. APC antigen-presenting cell, LAmB liposomal amphotericin B, MHC major histocompatibility complex, PDD peptide-derivatized-dendrimers, TH1 T-helper type 1
Dendrimers are a unique highly branched polymer with roughly spherical shape. They can be prepared by repetitive chemical reactions, adding layer after layer to their structures. Every new layer added to the periphery of a dendrimer is usually referred to as a ‘generation’. Fourth-generation and higher-generation dendrimers are macromolecules with high molecular weight and diameters in the nanometer range. Their chemistry can be controlled such that their surfaces are covered with functional groups that readily acquire positive charges, such as amines (−NH2), that are readily protonated (−NH3 +) at physiological pH. These macromolecules, with their highly positive charge, are ideal excipients with excellent solubility. A fraction of the surface amino groups on the dendrimer will remain unaltered and, therefore, will be available for positive charge development at physiological pH. The targeting moiety specific for macrophage/dendritic cells used in our studies is Pan DR Epitope (PADRE), a non-natural peptide that binds to the majority of major histocompatibility complex (MHC) class II molecules present on APCs such as monocytes, macrophages, dendritic cells, and Langerhans cells. PADRE binds to MHC class II from mice, rats, and humans. PADRE can perform two distinct functions: (1) as a ligand for MHC class II molecules, and (2) as a stimulator of adaptive immune responses. Thus far, PADRE has only been used for its in vivo T-helper stimulation in vaccination and immunotherapy. Here, for the first time, we are also using it for its ability to bind to MHC class II as a homing marker for APCs and infected macrophages.
A central idea in this nanocarrier concept is the derivatization of the dendrimer surface with a targeting peptide that will serve for homing to APCs [14••, 15•] (Fig. 1) and yet retain its immunopotentiating effect [14••, 15•]. Importantly, dendrimers can complex drugs via non-covalent interactions, such as electrostatic or hydrophobic interactions, which will then allow the drug to release in the cell cytosol and in low-pH environments. Positively charged nanocarriers can make electrostatic interactions with negatively charged drug cargo such as LAmB.
We used ethylenediamine core fifth-generation PAMAM dendrimers that contain 128 amine groups on their surfaces. These dendrimers maintain a low level of steric crowding on the dendrimer surface so that the amine groups maintain reactivity. The large number of amine groups on each dendrimer should afford (1) anchoring points for targeting functional groups, and (2) enough unaltered amine groups to maintain the positive charge on the surface required for strong electrostatic interactions with LAmB. A slightly altered version of a non-natural universal human leukocyte antigen (HLA)-DR-binding epitope (PADRE) [16•, 17•, 18], which has 16-amino acid peptide, binds with high- to intermediate-affinity to 15 of 16 of the most common HLA-DR types tested to date, and is thus capable of targeting all cells expressing such MHC class II molecules in mice and in the general human population. This PADRE is also able to activate CD4 T-helper cells and be immunogenic in both humans and mice [15•, 17•, 19, 16•, 20–22].
The amino terminus of the PADRE epitope is protected by acetylation, and the carboxylic acid of the terminal cysteine residue is used to achieve attachment via standard conjugation chemistry [14••, 15•]. We anticipated that attachment of several PADRE epitopes (two to eight) per dendrimer would enhance the targeting property of the drug- or DNA-delivery agents [15•], improving their properties for genetic vaccination purposes. However, it is important to leave a large number of unreacted amine groups so that the dendrimer will acquire a large positive charge via protonation at physiological pH values. Characterization of the PDD was done by ultraviolet–visible and fluorescence spectroscopy, elemental analysis, and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. A crucial structural issue that we must address is whether the PADRE peptide maintains a conformation suitable for binding, both in the PADRE-dendrimer conjugate and after complexation with LAmB. Binding activity was measured via flow cytometry as well as by circular dichroism (CD). These assays were used to monitor the spectra of the PADRE peptide before and after covalent attachment to the dendrimer.
In regards to using PDD for genetic vaccination, we are currently evaluating a number of novel gene-based adjuvants that are particularly appropriate for enhancing anti-leishmania immune responses. These adjuvants can be complexed to PDD as plasmids or RNA. One example is the chimeric molecule SPD-CD40L, which has been shown to enhance T-cell responses in vivo [23]. CD40L is a powerful inducer of macrophage and dendritic cell activation, and would be expected to increase antigen presentation and T-cell-mediated immune responses. Similarly, we have examined the use of the Epstein Barr Virus protein LMP1 as a gene-based adjuvant [24]. LMP1 is a molecular mimic of CD40 stimulation, and delivery of LMP1 expression plasmid to macrophages via PDD/LAmB should enhance macrophage control of infection and anti-leishmanial immunity. Finally, we have recent evidence that the chimeric molecules SPD-4-1BBL and SPD-BAFF are powerful inducers of T-cell-mediated immunity in mouse models [25]. Delivery of SPD-4-1BBL or SPD-BAFF expression plasmid by PDD-LAmB should enhance CD4+ and CD8+ T-cell responses against leishmania.
Conclusion
In vitro and in vivo assessment of PDD-LAmB showed enhanced efficacy of this novel immune-chemotherapy platform when compared with LAmB alone. In a mouse CL model, PDD-LAmB effectively targeted phagocytes and resulted in (1) ‘LAmB treatment reduction’ (PDD lowered the LAmB effective dose tenfold, shown in Fig. 2), (2) enhanced response kinetics, resulting in faster closure of lesions greater than 20 mm in diameter, (3) lowered toxicity as measured by MTT as well as blood chemistry, and, finally, (4) immune-enhancing effects that assisted the host in mounting anti-Leishmania-specific T-cell responses. Our ability to generate antigen-specific adaptive immunity without administering a vaccine is of great importance, and was associated with enhanced MHC class II expression in the draining lymph node, a sign of immune activation. It is likely that PDD shuttles the cargo (LAmB) into both macrophages and dendritic cells in vivo, resulting in more effective antigen uptake by dendritic cells that elicit anti-leishmania antigen-specific immune responses.

PDD accelerates the efficacy and lowers LAmB effective dose. Effects of different doses and formulation of LAmB on cutaneous leishmaniasis as assessed by lesion area. Mice bearing cutaneous Leishmania major lesions received either the full dose LAmB, or LAmB at a low dose (6.25 mg/K/day) either alone, encapsulated in a control nanoparticle (G5-Scr), or in PDD. PDD-engrafted LAmB has a significantly faster effect on reducing lesion areas. PDD2 is composed of a different universal helper peptide (APC ligand) and was coupled to PAMAM dendrimer. APC antigen-presenting cell, LAmB liposomal amphotericin B, PDD peptide-derivatized-dendrimers
It is of great concern that we may be getting close to a ‘post-antibiotic era’, and alternatives are urgently needed for the treatment of microbial infections. There is renewed interest in the use of immunotherapies with antibiotics or as stand-alone therapy to cure infectious diseases. We propose the use of PDD complexed with antibiotics as a novel method to combine antibiotics with immunotherapy. PDD uses a promiscuous T-helper epitope as the ‘APC-targeting moiety’, meaning that it bypasses MHC/HLA restriction. It is also of great importance to have a single product that can be used in both preclinical development (PDD binds APC of mice and monkeys) and in human trials (PDD binds all human APC). PDD-LAmB provides promising evidence that this approach is possible.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol. 2011;9(8):604–15.
Kara EE, Comerford I, Fenix KA, et al. Tailored immune responses: novel effector helper T cell subsets in protective immunity. PLoS Pathog. 2014;10(2):e1003905.
Guha R, Gupta D, Rastogi R, Vikram R, Krishnamurthy G, Bimal S, et al. Vaccination with leishmania hemoglobin receptor–encoding DNA protects against visceral leishmaniasis. Sci Transl Med. 2013;5(202):202ra121.
Schwarz TRK, Nahrendorf W, Masic A, Siewe L, Müller W, Roers A, et al. T cell-derived IL-10 determines leishmaniasis disease outcome and is suppressed by a dendritic cell-based vaccine. PLoS Pathog. 2013;9(6):e1003476.
Filardy AA, Costa-da-Silva AC, Koeller CM, et al. Infection with Leishmania major induces a cellular stress response in macrophages. PLoS One. 2014;9(1):e85715.
Alexander J, Bryson K. T helper (h)1/Th2 and Leishmania: paradox rather than paradigm. Immunol Lett. 2005;99(1):17–23.
Ricardo-Carter C, Favila M, Polando RE, et al. Leishmania major inhibits IL-12 in macrophages by signalling through CR3 (CD11b/CD18) and down-regulation of ETS-mediated transcription. Parasite Immunol. 2013;35(12):409–20.
Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3(8):711–5.
Santos DO, Coutinho CER, Madeira MF, et al. Leishmaniasis treatment–a challenge that remains: a review. Parasitol Res. 2008;103(1):1–10.
Adler-Moore J, Proffitt RT. Am Bisome: liposomal formulation, structure, mechanism of action and pre-clinical experience. J Antimicrob Chemother. 2002;49 Suppl 1:21–30.
McGwire BS, Satoskar AR. Leishmaniasis: clinical syndromes and treatment. QJM. 2014;107(1):7–14.
Chaubey P, Mishra B. Mannose-conjugated chitosan nanoparticles loaded with rifampicin for the treatment of visceral leishmaniasis. Carbohydr Polym. 2014;101:1101–8.
Utaile M, Kassahun A, Abebe T, Hailu A. Susceptibility of clinical isolates of Leishmania aethiopica to miltefosine, paromomycin, amphotericin B and sodium stibogluconate using amastigote-macrophage in vitro model. Exp Parasitol. 2013;134(1):68–75.
Daftarian PM, Stone GW, Kovalski L, et al. A targeted and adjuvanted nanocarrier lowers the effective dose of liposomal amphotericin B and enhances adaptive immunity in murine cutaneous leishmaniasis. J Infect Dis. 2013;208(11):1914–22. The first report on the use of PADRE for targeting drugs to APC via PADRE-derivatized-dendrimers.
Daftarian P, Kaifer AE, Li W, et al. Peptide-conjugated PAMAM dendrimer as a universal DNA vaccine platform to target antigen-presenting cells. Cancer Res. 2011;71(24):7452–62. The first report on a novel use of PADRE for targeting nucleic-acids to APC via PADRE-derivatized-dendrimers.
Daftarian P, Ali S, Sharan R, et al. Immunization with Th-CTL fusion peptide and cytosine-phosphate-guanine DNA in transgenic HLA-A2 mice induces recognition of HIV-infected T cells and clears vaccinia virus challenge. J Immunol. 2003;171(8):4028–39. PADRE enhances immunogenicity in vaccine setting in mice expressing human MHC II.
Daftarian P, Sharan R, Haq W, et al. Novel conjugates of epitope fusion peptides with CpG-ODN display enhanced immunogenicity and HIV recognition. Vaccine. 2005;23(26):3453–68. PADRE enhances immunogenicity in immunotherapeutic setting.
Daftarian P, Song GY, Ali S, et al. Two distinct pathways of immuno-modulation improve potency of p53 immunization in rejecting established tumors. Cancer Res. 2004;64(15):5407–14.
Alexander J, del Guercio MF, Maewal A, et al. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J Immunol. 2000;164(3):1625–33.
Daftarian P, Mansour M, Benoit AC, et al. Eradication of established HPV 16-expressing tumors by a single administration of a vaccine composed of a liposome-encapsulated CTL-T helper fusion peptide in a water-in-oil emulsion. Vaccine. 2006;24(24):5235–44.
Daftarian PM, Mansour M, Pohajdak B, et al. Rejection of large HPV-16 expressing tumors in aged mice by a single immunization of VacciMax encapsulated CTL/T helper peptides. J Transl Med. 2007;5:26.
Mansour M, Pohajdak B, Kast WM, et al. Therapy of established B16-F10 melanoma tumors by a single vaccination of CTL/T helper peptides in VacciMax. J Transl Med. 2007;5:20.
Stone GW, Barzee S, Snarsky V, et al. Multimeric soluble CD40 ligand and GITR ligand as adjuvants for human immunodeficiency virus DNA vaccines. J Virol. 2006;80(4):1762–72.
Gupta S, Termini JM, Niu L, et al. EBV LMP1, a viral mimic of CD40, activates dendritic cells and functions as a molecular adjuvant when incorporated into an HIV vaccine. J Leukoc Biol. 2011;90(2):389–98.
Kanagavelu S, Termini JM, Gupta S, et al. HIV-1 adenoviral vector vaccines expressing multi-trimeric BAFF and 4-1BBL enhance T cell mediated anti-viral immunity. PLoS One. 2014;9(2):e90100.
Compliance with Ethics Guidelines
Conflict of Interest
Pirouz M. Daftarian, Arba L. Ager, and Geoffrey Stone declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Daftarian, P.M., Ager, A.L. & Stone, G. A Targeted and Adjuvanted Nanoparticle for Immunochemotherapy of Leishmania Infections. Curr Trop Med Rep 1, 148–153 (2014). https://doi.org/10.1007/s40475-014-0023-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40475-014-0023-x