
Abstract
Purpose
Postoperative cognitive dysfunction (POCD) occurs frequently after cardiac surgery. The pathophysiology of POCD remains elusive, but previous work showed that intravenous lidocaine may be protective against POCD, possibly by modulating cerebral inflammation. We hypothesized that intravenous lidocaine would attenuate the cerebral inflammatory response to cardiopulmonary bypass (CPB) by reducing the transcerebral activation gradients of platelets, leukocytes, and/or platelet-leukocyte conjugates.
Methods
We studied 202 patients undergoing cardiac surgery with CPB in this prospective randomized double-blinded placebo-controlled trial. Subjects were randomized to receive either intravenous lidocaine (bolus + 48-hr infusion) or placebo (identical infusion volume and duration). Paired jugular venous and radial arterial blood samples were drawn at several time points and analyzed by fluorescence-activated cell sorting to identify activated platelets and platelet-leukocyte conjugates. Transcerebral activation gradients were calculated by subtracting arterial values from venous values and were compared between groups using repeated measures regression models with covariate adjustment for age, sex, surgery type, and CPB duration.
Results
Beginning after aortic cross-clamp release and peaking ten minutes after the termination of CPB, the mean (SD) transcerebral activation gradient of platelet-monocyte conjugates decreased in lidocaine-treated vs placebo-treated patients [−1.84 (11.47) mean linear fluorescence intensity (MLFI) vs 1.46 (13.88) MLFI, respectively; mean difference, −4.08 MLFI; 95% confidence interval, −7.86 to −0.29; P = 0.03). No difference was seen at any time point for activated platelets or for platelet-neutrophil conjugates.
Conclusion
While lidocaine did not affect the systemic or transcerebral activation of platelets or leukocytes, we did observe a reduction in the transcerebral activation of platelet-monocyte conjugates after aortic cross-clamp release. This may be a manifestation of reduced cerebral inflammation during cardiopulmonary bypass in response to treatment with lidocaine. This trial was registered at ClinicalTrials.gov (NCT00938964).
Résumé
Objectif
Le dysfonctionnement cognitif postopératoire (DCPO) constitue une complication fréquente après une chirurgie cardiaque. La physiopathologie du DCPO est encore peu connue, mais des travaux réalisés par le passé ont démontré que la lidocaïne intraveineuse pourrait avoir un effet protecteur contre le DCPO, possiblement en modulant l’inflammation cérébrale. Nous avons émis l’hypothèse que la lidocaïne intraveineuse atténuerait la réponse inflammatoire cérébrale à la circulation extracorporelle (CEC) en réduisant les gradients d’activation transcérébrale des plaquettes, des leucocytes, et/ou des conjugués plaquettes-leucocytes.
Méthode
Nous avons étudié 202 patients subissant une chirurgie cardiaque avec CEC dans cette étude prospective randomisée à double insu et contrôlée par placebo. Les patients ont été randomisés à recevoir de la lidocaïne intraveineuse (bolus + perfusion de 48 h) ou un placebo (volume et durée de perfusion identiques). Des échantillons de sang appariés de la veine jugulaire et de l’artère radiale ont été prélevés à différents moments dans le temps et analysés par tri de cellules activées par fluorescence afin d’identifier les plaquettes et les conjugués plaquettes-leucocytes activés. Les gradients d’activation transcérébrale ont été calculés en soustrayant les valeurs artérielles aux valeurs veineuses puis comparés entre les groupes à l’aide de modèles de régression à mesures répétées avec ajustement des covariables pour tenir compte de l’âge, du sexe, du type de chirurgie et de la durée de la CEC.
Résultats
En commençant à la fin du clampage aortique et atteignant son pic dix minutes après la fin de la CEC, le gradient d’activation transcérébrale moyen (ÉT) des conjugués plaquettes-monocytes a baissé dans le groupe de patients traités avec de la lidocaïne par rapport au groupe placebo [−1,84 (11,47) intensité de fluorescence linéaire moyenne (IFLM) vs 1,46 (13,88) IFLM, respectivement; différence moyenne, −4,08 IFLM; intervalle de confiance 95 %, −7,86 à −0,29; P = 0,03). Aucune différence n’a été observée à quelque moment dans le temps que ce soit en ce qui touchait aux plaquettes activées et aux conjugués plaquettes-neutrophiles.
Conclusion
Bien que la lidocaïne n’ait pas eu d’impact sur l’activation systémique ou transcérébrale des plaquettes ou des leucocytes, nous avons observé une réduction de l’activation transcérébrale des conjugués plaquettes-monocytes après la fin du clampage aortique. Il pourrait s’agir d’une manifestation de la réduction de l’inflammation cérébrale pendant la circulation extracorporelle en réponse au traitement à base de lidocaïne. Cette étude a été enregistrée au ClinicalTrials.gov (NCT00938964).
Similar content being viewed by others
Cognitive dysfunction after cardiac surgery with cardiopulmonary bypass (CPB) occurs in up to 50% of patients at hospital discharge and can persist in nearly one-third of patients at six weeks after surgery.1,2 Postoperative cognitive dysfunction (POCD) can lead not only to prolonged hospitalization but also to decreased patient quality of life after hospital discharge.3,4 The mechanism(s) of POCD remains elusive, although several contributing factors have been proposed,1 including the exacerbation of preoperative cognitive impairment,5 genetic predisposition, cerebral embolism/hypoperfusion, hemodilution, hyperglycemia, anesthetic neurotoxicity,6–8 and perioperative hyperthermia.9 In addition to these mechanisms, cerebral inflammation in response to embolic and other events during CPB, manifested by transcerebral platelet activation,10 has also been proposed as a mechanism linking POCD to cardiac surgery.10,11 Given the mechanistic uncertainty behind POCD, it is not surprising that few interventions to date have been successful in ameliorating POCD in patients undergoing cardiac surgery.12–14
Several studies have reported the beneficial effects of lidocaine, an antiarrhythmic sodium-channel blocker, on POCD after both cardiac and non-cardiac surgery.15–17 Indeed, our own previous work has suggested that low-dose intravenous lidocaine is protective against POCD in non-diabetic cardiac surgical patients.18 The mechanism for this protective effect of lidocaine may be related to a cerebral anti-inflammatory effect as lidocaine has previously been shown to attenuate systemic inflammatory responses.19–23 Specifically, lidocaine reduces granulocyte adherence to injured endothelium at sites of inflammation,24 reduces leukocyte adherence during endotoxemia,25 and decreases neutrophil adhesion molecule expression during early reperfusion after tourniquet-induced ischemia.26
Given the previously demonstrated association between transcerebral platelet activation and POCD10 and the inflammatory modulating properties of lidocaine on multiple inflammatory cell types, we planned a substudy of a larger ongoing trial investigating the effect of lidocaine on POCD and hypothesized a priori that lidocaine would attenuate the cerebral inflammatory response during CPB. We predicted that this would manifest by a reduction in the transcerebral activation gradients of platelets (platelet surface P-selectin expression),27 leukocytes (increased monocyte and neutrophil surface CD11b expression),28 and/or platelet-leukocyte conjugates (colocalization of CD11b and the general platelet marker CD41).
Methods
Study population
In May 2009, the Duke University Health System Institutional Review Board approved this substudy which was part of an ongoing larger (planned enrolment: 476 patients) prospective randomized double-blinded placebo-controlled clinical trial evaluating the effects of intravenous lidocaine on neurocognitive outcomes after cardiac surgery with CPB (clinicaltrials.gov; NCT00938964).
Following written informed consent, 202 patients scheduled to undergo coronary artery bypass grafting (CABG), with or without concomitant valve surgery, or valve surgery alone and utilizing CPB were enrolled into this substudy. Exclusion criteria included planned circulatory arrest, history of symptomatic cerebrovascular disease (e.g., stroke with residual deficit), psychiatric illness (e.g., any clinical diagnosis requiring therapy), renal failure (serum creatinine > 2 mg·dL−1), liver failure (aspartate aminotransferase and/or alanine aminotransferase >1.5x upper limit of normal), consumption of more than two alcoholic beverages per day, inability to read, less than a seventh-grade education, and a baseline Mini-Mental State Examination score of < 24.
Subjects were randomized to receive one of two treatments: 1) intravenous lidocaine bolus (1 mg·kg−1) on induction, followed by a continuous infusion of 48 µg·kg−1·min−1 for one hour, 24 µg·kg−1·min−1 for the second hour, and 10 µg·kg−1·min−1 for the subsequent 46 hr29 or 2) placebo, normal saline given as an identical volume bolus and infusion, with rate changes as in the treatment group to preserve blinding. The study statistician prepared a group assignment schedule using a randomization function in SAS® (Statistical Analysis System; SAS Institute, Inc., Cary, NC, USA), and patients’ assignments were stored in consecutively numbered sealed envelopes until allocation. Randomization was performed prior to surgery and after confirmation of the procedure type and planned use of CPB. The patients, all care providers, and study personnel were blinded to the treatment assignment. Since the parent study is ongoing, we preserved blinding by allowing the substudy statistician access to the randomization schedule pertaining only to patients enrolled in the substudy. Furthermore, all of the patients in this substudy completed their final follow-up for the parent trial prior to the analysis of the data for this substudy.
Patient management
Anesthesia was induced with midazolam and fentanyl, and isoflurane was used for maintenance. All patients underwent nonpulsatile hypothermic (30-32°C) CPB with a membrane oxygenator and 32-μm arterial line filter by means of a pump primed with crystalloid. Serial hematocrit levels were maintained at ≥ 0.21. Before initiating CPB, heparinization (300-400 U·kg−1) was performed to a target activated coagulation time (ACT) of > 480 sec. Perfusion was maintained at flow rates of 2-2.4 L·min−1·m−2 throughout CPB to maintain a mean arterial pressure of 50-80 mmHg. Arterial blood gases were measured every 15-30 min to maintain the PaCO2 at 35-40 mmHg unadjusted for temperature and the PaO2 at 150-250 mmHg. During surgery, all patients received a bolus and infusion of a lysine analogue antifibrinolytic agent (either epsilon-aminocaproic acid as a bolus of 10 g followed by an infusion of 1 g·hr−1 or tranexamic acid as a bolus of 1 mg·kg−1 followed by an infusion of 1 mg·kg−1·hr−1). Following termination of CPB, protamine was administered at a dose of 0.5-1 mg/100 units of initial heparin dose, with additional doses guided by ACT, activated partial thromboplastin time (aPTT), or heparin level. In general, target hemoglobin was 8-9 g·dL−1, and coagulopathic bleeding was treated with plasma, platelets, or cryoprecipitate guided by standard laboratory and viscoelastic testing, as recommended in the Society of Cardiovascular Anesthesiologists and Society of Thoracic Surgeons guidelines.30
Blood sampling
Paired whole blood samples were drawn from the radial artery and jugular vein at four time points: 1) baseline (prior to procedure), 2) five minutes after release of the aortic cross-clamp (XCR), 3) ten minutes after the termination of CPB (endCPB), and 4) six hours after release of the aortic cross-clamp (6h pXCR). The blood was immediately fixed with 1% paraformaldehyde in phosphate-buffered saline solution.
Reagents and flow cytometry
Whole blood samples were fixed, washed, and labelled with monoclonal antibodies as previously described.31 Platelets and leukocytes were identified using fluorochrome-conjugated monoclonal antibodies for platelet-specific monoclonal antibodies [anti-CD41 (glycoprotein IIb/IIIa) and anti-CD62P (P-selectin)] and leukocyte-specific monoclonal antibodies [anti-CD11b (integrin alpha M)] (Becton Dickinson Biosciences, San Jose, CA, USA). Activated platelets were identified by the expression of surface P-selectin. Platelet-monocyte and platelet-neutrophil conjugates were identified by the colocalization of CD11b and the general platelet marker CD41. Monocytes and neutrophils were differentiated by side-scatter clustering. Samples were analyzed by fluorescence-activated cell sorting (FACS) on a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA). Marker expression was quantified as mean linear fluorescence intensity (MLFI), which is proportional to platelet activation and platelet-leukocyte binding.32
Statistical methods
A power analysis based on our previous study,10 which found a mean (standard deviation [SD]) platelet activation gradient of 9.3 (12.9) MLFI, estimated that 200 patients would provide 90% power at alpha 0.05 to detect a difference of 5.2% (effect size of 0.46 MLFI) in platelet activation between groups. This represents the difference in platelet activation observed in our previous study between patients who did and did not develop POCD. With no in vivo a priori data specifically related to the effects of lidocaine on leukocytes or platelet-leukocyte conjugate formation,33 we assumed the same sample size as for platelet activation for all of our proposed outcomes.
Categorical and continuous demographic data were compared between the groups with χ2 test, Wilcoxon rank-sum test, or Student’s t test, as appropriate. Descriptive summaries are presented as counts and proportions for categorical variables and mean (SD) for continuous variables.
The primary outcomes were the transcerebral gradients of activated platelets and platelet-leukocyte conjugates. Platelet-leukocyte conjugates were chosen because they are known to be more sensitive and longer-lived markers of in vivo platelet activation than surface P-selectin expression.32,34,35 Secondary outcomes were the transcerebral gradients of activated monocytes and neutrophils. The transcerebral gradient was determined by subtracting the arterial value from the corresponding jugular venous value. To derive adjusted estimates of group differences at each point in the time course of transcerebral gradients, we used repeated measures regression models with covariate adjustment for age, sex, surgery type, and CPB time—factors thought to influence the risk of POCD. The normality of the Studentized model residuals was assessed with Q-Q plots and kernel densities. The estimated least-squares means of the resulting model were compared at each time point with two-sided Student’s t tests. All analyses were performed with SAS® version 9.4 (SAS Institute Inc.; Cary, NC, USA). All reported P values are two sided.
Results
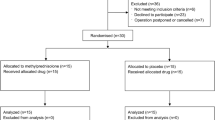
A CONSORT (Consolidated Standards of Reporting Trials) diagram detailing the recruitment and analysis of patients is presented in Fig. 1. Patient demographics and intraoperative details are summarized in Table 1. The lidocaine and placebo groups were similar with respect to demographic characteristics. Nevertheless, the placebo group did have more CABG procedures compared with the lidocaine group, so we accounted for surgical procedure type in the subsequent statistical modelling.

CONSORT diagram showing the flow of study participants
Our primary endpoint of transcerebral platelet activation gradient, as defined by expression of the short-lived platelet surface marker P-selectin, did not differ between placebo and treatment groups at any time point. We also did not observe a difference in the transcerebral leukocyte activation gradient (i.e., increased neutrophil and monocyte CD11b expression over baseline) at any time point. Finally, we did not detect a difference in the transcerebral activation gradient for platelet-neutrophil conjugates between control and lidocaine-treated patients (Table 2).
The transcerebral gradient of platelet-monocyte conjugates decreased in the lidocaine group beginning at XCR and was significantly different from placebo at endCPB (Fig. 2). In the repeated measures analysis adjusting for covariates, the mean (SD) transcerebral activation gradient of platelet-monocyte conjugates decreased in lidocaine-treated vs placebo-treated patients [−1.84 (11.47) MLFI vs 1.46 (13.88) MLFI, respectively; mean difference, −4.08 MLFI; 95% confidence interval, −7.86 to −0.29; P = 0.03]. Comparing the arterial (Fig. 3, panel A) and jugular venous (Fig. 3, panel B) values of the platelet-monocyte conjugates, the arterial levels (representing systemic inflammation, as the sample was from the radial artery) of platelet-monocyte conjugate formation did not differ between treatment groups. The observed reduction in the transcerebral platelet-monocyte gradient in patients who received lidocaine was almost entirely determined by a decrease in the jugular venous (i.e., potentially cerebral inflammatory) levels.

Transcerebral (jugular-arterial) gradient of platelet-monocyte conjugates at baseline, five minutes after release of the aortic cross-clamp (XCR), ten minutes after the termination of cardiopulmonary bypass (EndCPB), and six hours after release of the aortic cross-clamp (6h pXCR) in cardiac surgical patients treated with placebo vs intravenous lidocaine. Error bars denote standard error of the mean. *P = 0.03

Arterial (A) and jugular (B) levels of platelet-monocyte conjugates at baseline, five minutes after release of the aortic cross-clamp (XCR), ten minutes after the termination of cardiopulmonary bypass (EndCPB), and six hours after release of the aortic cross-clamp (6h pXCR) in cardiac surgical patients treated with placebo vs intravenous lidocaine. Error bars denote standard error of the mean
Discussion
In this prospective double-blind placebo-controlled randomized clinical trial of intravenous lidocaine administered during cardiac surgery with CPB, we found that lidocaine did not alter the transcerebral gradient of activated platelets or leukocytes. Nevertheless, we did find a reduced transcerebral gradient of platelet-monocyte conjugates in lidocaine-treated patients. This reduction began at aortic cross-clamp release, peaked ten minutes after the termination of CPB, and returned to baseline by six hours post-reperfusion. This reduction in the transcerebral gradient was primarily the result of a decreased efflux of these conjugates from the cerebral circulation. This decrease in platelet-monocyte conjugate activation in the brain may be a manifestation of reduced transcerebral inflammation during cardiopulmonary bypass in response to treatment with lidocaine.
Lidocaine is a class IB antiarrhythmic agent that readily crosses the blood-brain barrier and confers cerebral protection in both surgical and non-surgical patient settings where inflammation may be the predominate mechanism of injury.18,36 Lidocaine has been shown to inhibit multiple inflammatory responses, including inhibition of leukotriene and interleukin-1α release from polymorphonuclear leukocytes (PMNs).37 This produces morphologic changes that inhibit PMN adherence38 and thereby reduce PMN delivery to sites of inflammation, inhibit PMN migration and accumulation at sites of inflammation,24,39,40 and block the priming of neutrophils, a potentiation response to several agents that has been implicated in the overstimulation of inflammatory pathways leading to undesirable damage to host tissue.20,41 While the effects of local anesthetics on neutrophils and macrophages have been previously characterized, we present a novel finding of an attenuation in platelet-monocyte conjugates in cardiac surgical patients. Interestingly, we did not find any change in the systemic (i.e., radial arterial) levels of any of the measured inflammatory cells between patients treated with lidocaine vs placebo. Assuming that transcerebral inflammatory activation is largely a result of embolic phenomenon, such as that associated with surgical manipulation of the aorta, and thus may be more intensely seen in the cerebral circulation than in the systemic circulation, this may explain why we observed differential effects of lidocaine on the attenuation of inflammatory cells between the cerebral and systemic vascular compartments.
Transcerebral inflammation, more specifically transcerebral platelet activation, has been previously shown to correlate with POCD in patients after cardiac surgery with CPB.10 In that study, the largest increase in the transcerebral platelet activation gradient occurred five minutes after aortic cross-clamp release, suggesting that atheromatous or gaseous emboli may trigger cerebral inflammation during cardiac surgery. The time course is not surprising, given that the vast majority (> 80%) of measurable microemboli are generated at the point of aortic cross-clamp release.42 Although we did not observe a difference in activated platelets between the placebo and lidocaine groups in our current study, we did observe a significant difference in platelet-monocyte conjugate levels. P-selectin is a platelet marker that is rapidly expressed but is also rapidly internalized, accounting for its transient expression on activated platelets.43 In contrast, platelet-leukocyte conjugates are formed more gradually over a period of up to 15 min as the result of the binding of P-selectin-positive platelets to neutrophils or monocytes. These conjugates are thus viewed as longer-lived markers of platelet activation.32,34 The level of platelet-monocyte conjugates, in particular, has been shown to be a more sensitive indicator of in vivo platelet activation than P-selectin expression.34 Platelet-monocyte conjugate levels in the blood are significantly increased in association with macrovascular ischemic events44,45 and in response to microcirculatory injury.46 Previous work has shown that a plaque-derived lipid (specifically lysophosphatidic acid), which would likely be released into the circulation during plaque disruption with aortic cross-clamp manipulation, stimulates platelet activation and subsequent platelet-monocyte conjugate formation.47 In the current study, the transcerebral formation of platelet-monocyte conjugates in the lidocaine-treated patients was reduced beginning immediately after aortic cross-clamp release and peaked significantly at ten minutes after CPB termination,. This suggests that the cerebral inflammatory response to atheromatous emboli to the brain released during aortic cross-clamp manipulation is reduced in lidocaine-treated patients when compared with controls.
Our finding that lidocaine can reduce the formation of platelet-monocyte conjugates in the cerebral vascular bed during cardiac surgery has been previously shown in in vitro studies. Huang et al. reported that lidocaine mixed with whole blood inhibited platelet P-selectin expression and platelet-leukocyte conjugate formation in response to lipopolysaccharide stimulation.33 Although these investigators used supratherapeutic lidocaine concentrations, this study supports the finding that lidocaine can modulate platelet activation and platelet-leukocyte conjugate formation under inflammatory conditions.
It remains unknown if platelet-monocyte conjugates are merely associated with cerebrovascular inflammation or whether they are actively participating in inflammatory-mediated cerebral injury. The formation of platelet-leukocyte conjugates has long been associated with vascular disease in several conditions, including congestive heart failure, stroke, acute coronary syndromes, diabetes, and hypertension,48 and following balloon angioplasty.34 Platelet aggregation with monocytes increases the adhesive properties of monocytes to the endothelium and stimulates tissue factor expression on monocytes that promotes fibrin formation and may accelerate the formation of intravascular thrombosis.49,50 Not surprisingly, platelet-monocyte conjugates are an early marker of acute myocardial infarction.51 Platelet-monocyte conjugates have also been implicated in the progression of established arteriosclerosis, thrombosis, and inflammation via local cytokine-mediated recruitment of additional leukocytes and platelets.48 Finally, platelet-monocyte conjugate formation stimulates monokine synthesis, which produces an imbalanced proinflammatory response.52,53
Previous work by our group has shown increased transcerebral platelet activation in patients who suffered neurocognitive injury during cardiac surgery with CPB,10 which raises several questions in light of our current findings. First, we did not observe any difference in platelet activation as measured by P-selectin expression between the treatment groups in this study. This may be related to the surgical case mix of our current study compared with the previous study. In contrast to the previous study, which evaluated only those patients undergoing CABG, our current study included a significant proportion of patients who underwent CABG with concomitant valve surgery or valve surgery alone (Table 1). Work by Leguyader et al. has shown that platelet-monocyte conjugate formation, but not platelet P-selectin expression, was seen in patients who underwent valve replacement with a bioprosthesis.54 Second, although platelet-leukocyte conjugates are generally markers of platelet activation, in this study, we detected a difference only in platelet-monocyte conjugates and not in platelet-neutrophil conjugates. Even though both platelet-neutrophil and platelet-monocyte conjugates are formed in response to platelet activation in vitro, evidence suggests that in vivo platelet-monocyte conjugates are preferentially formed. This is due to the fact that the leukocyte receptor PSGL-1 (P-selectin glycoprotein ligand-1), to which platelets bind via P-selectin, is expressed in vivo at a markedly higher density on monocytes than on neutrophils.55 Thus, in vivo platelet-monocyte conjugates comprise the overwhelming majority of platelet-leukocyte conjugates formed in response to platelet activation.
A limitation of the current study is that the neurocognitive outcomes data on the patients treated with lidocaine vs placebo are not yet available as the parent trial is ongoing. Until we have data on the short- and long-term cognitive outcomes between the groups, we will not be able to assess the clinical relevance of our findings. Nevertheless, if lidocaine proves to be protective against POCD in this large clinical trial, as was suggested by previous work,18 these results may offer a mechanistic explanation for such a neurocognitive benefit in patients undergoing cardiac surgery with CPB. Second, despite the evidence in the literature to suggest an inflammatory modulating effect of lidocaine, there were insufficient data related to effects on platelet-leukocyte conjugates to suggest a priori an appropriate study sample size. Based on the effect size observed in the current study (0.31), we estimate that a future study would require 480 patients (240 per treatment group) to have 80% power to confirm the findings observed at endCPB with an adjusted alpha of 0.0125. As a result, we acknowledge that the current study is underpowered to make any definitive conclusions regarding the effects of lidocaine on the inflammatory cells examined. Finally, there is a lack of clear evidence in the literature to suggest which inflammatory cell type(s) is the most reliable or superior marker of cerebral inflammatory injury. While previous work has shown an association between activated platelets in the cerebral circulation and cerebral injury (i.e., POCD),10 our current study found no effect of lidocaine on platelet activation. This may not be surprising given that the evidence for an inflammatory modulating effect of lidocaine has so far been predominantly documented for nucleated inflammatory cells, in particular neutrophils.20 Regardless, our finding of a difference in platelet-monocyte conjugates in the cerebral circulation between patients who received lidocaine vs placebo indicates that cell types other than platelets may be involved in cerebral inflammatory injury and should be considered in future investigations.
In summary, we have shown that lidocaine treatment reduced the transcerebral formation of platelet-monocyte conjugates in patients undergoing cardiac surgery, specifically after aortic cross-clamp release and the termination of CPB. These findings point to a potential inflammatory modulating property of lidocaine in the cerebral vascular bed during cardiac surgery that merits validation in future studies.
References
Hogue CW Jr, Palin CA, Arrowsmith JE. Cardiopulmonary bypass management and neurologic outcomes: an evidence-based appraisal of current practices. Anesth Analg 2006; 103: 21-37.
Newman MF, Kirchner JL, Phillips-Bute B, et al. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med 2001; 344: 395-402.
Newman MF, Grocott HP, Mathew JP, et al. Report of the substudy assessing the impact of neurocognitive function on quality of life 5 years after cardiac surgery. Stroke 2001; 32: 2874-81.
Phillips-Bute B, Mathew JP, Blumenthal JA, et al. Association of neurocognitive function and quality of life 1 year after coronary artery bypass graft (CABG) surgery. Psychosom Med 2006; 68: 369-75.
Silbert B, Evered L, Scott DA, et al. Preexisting cognitive impairment is associated with postoperative cognitive dysfunction after hip joint replacement surgery. Anesthesiology 2015; 122: 1224-34.
Chan MT, Cheng BC, Lee TM, Gin T, CODA Trial Group. BIS-guided anesthesia decreases postoperative delirium and cognitive decline. J Neurosurg Anesthesiol 2013; 25: 33-42.
Radtke FM, Franck M, Lendner J, Kruger S, Wernecke KD, Spies CD. Monitoring depth of anaesthesia in a randomized trial decreases the rate of postoperative delirium but not postoperative cognitive dysfunction. Br J Anaesth 2013; 110(Suppl 1): i98-105.
Berger M, Nadler JW, Browndyke J, et al. Postoperative cognitive dysfunction: minding the gaps in our knowledge of a common postoperative complication in the elderly. Anesthesiol Clin 2015; 33: 517-50.
Grocott HP, Mackensen GB, Grigore AM, et al. Postoperative hyperthermia is associated with cognitive dysfunction after coronary artery bypass graft surgery. Stroke 2002; 33: 537-41.
Mathew JP, Rinder HM, Smith BR, Newman MF, Rinder CS. Transcerebral platelet activation after aortic cross-clamp release is linked to neurocognitive decline. Ann Thorac Surg 2006; 81: 1644-9.
Mathew JP, Podgoreanu MV, Grocott HP, et al. Genetic variants in P-selectin and C-reactive protein influence susceptibility to cognitive decline after cardiac surgery. J Am Coll Cardiol 2007; 49: 1934-42.
Mathew JP, Shernan SK, White WD, et al. Preliminary report of the effects of complement suppression with pexelizumab on neurocognitive decline after coronary artery bypass graft surgery. Stroke 2004; 35: 2335-9.
Doraiswamy PM, Babyak MA, Hennig T, et al. Donepezil for cognitive decline following coronary artery bypass surgery: a pilot randomized controlled trial. Psychopharmacol Bull 2007; 40: 54-62.
Grigore AM, Mathew J, Grocott HP, et al. Prospective randomized trial of normothermic versus hypothermic cardiopulmonary bypass on cognitive function after coronary artery bypass graft surgery. Anesthesiology 2001; 95: 1110-9.
Mitchell SJ, Pellett O, Gorman DF. Cerebral protection by lidocaine during cardiac operations. Ann Thorac Surg 1999; 67: 1117-24.
Chen K, Wei P, Zheng Q, Zhou J, Li J. Neuroprotective effects of intravenous lidocaine on early postoperative cognitive dysfunction in elderly patients following spine surgery. Med Sci Monit 2015; 21: 1402-7.
Wang D, Wu X, Li J, Xiao F, Liu X, Meng M. The effect of lidocaine on early postoperative cognitive dysfunction after coronary artery bypass surgery. Anesth Analg 2002; 95: 1134-41.
Mathew JP, Mackensen GB, Phillips-Bute B, et al. Randomized, double-blinded, placebo controlled study of neuroprotection with lidocaine in cardiac surgery. Stroke 2009; 40: 880-7.
Hollmann MW, Gross A, Jelacin N, Durieux ME. Local anesthetic effects on priming and activation of human neutrophils. Anesthesiology 2001; 95: 113-22.
Hollmann MW, Durieux ME. Local anesthetics and the inflammatory response: a new therapeutic indication? Anesthesiology 2000; 93: 858-75.
Picardi S, Cartellieri S, Groves D, et al. Local anesthetic-induced inhibition of human neutrophil priming: the influence of structure, lipophilicity, and charge. Reg Anesth Pain Med 2013; 38: 9-15.
Condliffe AM, Kitchen E, Chilvers ER. Neutrophil priming: pathophysiological consequences and underlying mechanisms. Clin Sci (Lond) 1998; 94: 461-71.
Liu J, Zhang H, Qi Z, Zheng X. Lidocaine protects against renal and hepatic dysfunction in septic rats via downregulation of Toll-like receptor 4. Mol Med Rep 2014; 9: 118-24.
MacGregor RR, Thorner RE, Wright DM. Lidocaine inhibits granulocyte adherence and prevents granulocyte delivery to inflammatory sites. Blood 1980; 56: 203-9.
Schmidt W, Schmidt H, Bauer H, Gebhard MM, Martin E. Influence of lidocaine on endotoxin-induced leukocyte-endothelial cell adhesion and macromolecular leakage in vivo. Anesthesiology 1997; 87: 617-24.
Lan W, Harmon D, Wang JH, Ghori K, Shorten G, Redmond P. The effect of lidocaine on in vitro neutrophil and endothelial adhesion molecule expression induced by plasma obtained during tourniquet-induced ischaemia and reperfusion. Eur J Anaesthesiol 2004; 21: 892-7.
Michelson AD, Furman MI. Laboratory markers of platelet activation and their clinical significance. Curr Opin Hematol 1999; 6: 342-8.
Springer TA. Adhesion receptors of the immune system. Nature 1990; 346: 425-34.
Hsu YW, Somma J, Newman MF, Mathew JP. Population pharmacokinetics of lidocaine administered during and after cardiac surgery. J Cardiothorac Vasc Anesth 2011; 25: 931-6.
Society of Thoracic Surgeons Blood Conservation Guideline Task Force; Ferraris VA, Brown JR, Despotis GJ, et al. 2011 update to the Society of Thoracic Surgeons and the Society of Cardiovascular Anesthesiologists blood conservation clinical practice guidelines. Ann Thorac Surg 2011; 91: 944-82.
Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Dynamics of leukocyte-platelet adhesion in whole blood. Blood 1991; 78: 1730-7.
Rinder CS, Bonan JL, Rinder HM, Mathew J, Hines R, Smith BR. Cardiopulmonary bypass induces leukocyte-platelet adhesion. Blood 1992; 79: 1201-5.
Huang GS, Lin TC, Wang JY, Ku CH, Ho ST, Li CY. Lidocaine priming reduces ADP-induced P-selectin expression and platelet-leukocyte aggregation. Acta Anaesthesiol Taiwan 2009; 47: 56-61.
Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001; 104: 1533-7.
Flierl U, Bauersachs J, Schafer A. Modulation of platelet and monocyte function by the chemokine fractalkine (CX3 CL1) in cardiovascular disease. Eur J Clin Invest 2015; 45: 624-33.
Mitchell SJ. Lidocaine in the treatment of decompression illness: a review of the literature. Undersea Hyperb Med 2001; 28: 165-74.
Sinclair R, Eriksson AS, Gretzer C, Cassuto J, Thomsen P. Inhibitory effects of amide local anaesthetics on stimulus-induced human leukocyte metabolic activation, LTB4 release and IL-1 secretion in vitro. Acta Anaesthesiol Scand 1993; 37: 159-65.
Rabinovitch M, DeStefano MJ. Cell shape changes induced by cationic anesthetics. J Exp Med 1976; 143: 290-304.
Hammer R, Dahlgren C, Stendahl O. Inhibition of human leukocyte metabolism and random mobility by local anaesthesia. Acta Anaesthesiol Scand 1985; 29: 520-3.
Eriksson AS, Sinclair R, Cassuto J, Thomsen P. Influence of lidocaine on leukocyte function in the surgical wound. Anesthesiology 1992; 77: 74-8.
Fischer LG, Bremer M, Coleman EJ, et al. Local anesthetics attenuate lysophosphatidic acid-induced priming in human neutrophils. Anesth Analg 2001; 92: 1041-7.
Reinsfelt B, Ricksten SE, Zetterberg H, Blennow K, Freden-Lindqvist J, Westerlind A. Cerebrospinal fluid markers of brain injury, inflammation, and blood-brain barrier dysfunction in cardiac surgery. Ann Thorac Surg 2012; 94: 549-55.
Kansas GS. Selectins and their ligands: current concepts and controversies. Blood 1996; 88: 3259-87.
Lippi G, Montagnana M, Salvagno GL, et al. Risk stratification of patients with acute myocardial infarction by quantification of circulating monocyte-platelet aggregates. Int J Cardiol 2007; 115: 101-2.
McCabe DJ, Harrison P, Mackie IJ, et al. Platelet degranulation and monocyte-platelet complex formation are increased in the acute and convalescent phases after ischaemic stroke or transient ischaemic attack. Br J Haematol 2004; 125: 777-87.
Elalamy I, Chakroun T, Gerotziafas GT, et al. Circulating platelet-leukocyte aggregates: a marker of microvascular injury in diabetic patients. Thromb Res 2008; 121: 843-8.
Haseriuck N, Erl W, Pandey D, et al. The plaque lipid lysophosphatidic acid stimulates platelet activation and platelet-monocyte aggregate formation in whole blood: involvement of P2Y1 and P2Y12 receptors. Blood 2004; 103: 2585-92.
da Costa Martins P, Zwaginga JJ. Leukocyte-platelet aggregates: new particles reflecting and effecting cardiovascular disease. Thromb Haemost 2005; 94: 1120-1.
Celi A, Pellegrini G, Lorenzet R, et al. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci USA 1994; 91: 8767-71.
Kirchhofer D, Riederer MA, Baumgartner HR. Specific accumulation of circulating monocytes and polymorphonuclear leukocytes on platelet thrombi in a vascular injury model. Blood 1997; 89: 1270-8.
Furman MI, Barnard MR, Krueger LA, et al. Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol 2001; 38: 1002-6.
Weyrich AS, Elstad MR, McEver RP, et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest 1996; 97: 1525-34.
Rondina MT, Carlisle M, Fraughton T, et al. Platelet-monocyte aggregate formation and mortality risk in older patients with severe sepsis and septic shock. J Gerontol A Biol Sci Med Sci 2015; 70: 225-31.
Leguyader A, Watanabe R, Berbe J, Boumediene A, Cogne M, Laskar M. Platelet activation after aortic prosthetic valve surgery. Interact Cardiovasc Thorac Surg 2006; 5: 60-4.
Kappelmayer J, Kiss A, Karaszi E, Veszpremi A, Jako J, Kiss C. Identification of P-selectin glycoprotein ligand-1 as a useful marker in acute myeloid leukaemias. Br J Haematol 2001; 115: 903-9.
Funding
This study was supported by grants to Dr. Mathew from the National Institutes of Health (HL096978, HL108280, HL109971).
Conflicts of interest
None declared.
Author contributions
Rebecca Y. Klinger, Joseph P. Mathew, and Thomas L. Ortel contributed substantially to the acquisition of data. Rebecca Y. Klinger and Joseph P. Mathew contributed substantially to the analysis of data and drafting the article. Mary Cooter contributed to the analysis of data. Rebecca Y. Klinger, Joseph P. Mathew, Mary Cooter, and Thomas L. Ortel contributed substantially to the interpretation of data. Rebecca Y. Klinger, Joseph P. Mathew, Miles Berger, Mihai V. Podgoreanu, Mark Stafford-Smith, Ian J. Welsby, Jerrold H. Levy, Henry M. Rinder, Mark F. Newman, and Thomas L. Ortel contributed substantially to the conception and design of the manuscript.
Editorial responsibility
This submission was handled by Dr. Hilary P. Grocott, Editor-in-Chief, Canadian Journal of Anesthesia.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
A list of all members of the Neurologic Outcome Research Group is given in the Appendix.
Appendix
Appendix
Neurologic Outcome Research Group of the Duke Heart Center
Director: Joseph P. Mathew MD, Co-Director: James A. Blumenthal PhD.
Anesthesiology: Miles Berger MD, PhD, Miklos D. Kertai MD, Yi-Ju Li PhD, Frederick W. Lombard MD, Joseph P. Mathew MD, Mark F. Newman MD, Mihai V. Podgoreanu MD, Mark Stafford-Smith MD, Madhav Swaminathan MD, Niccolo Terrando PhD, David S. Warner MD, Bonita L. Funk RN, CCRP, Narai Balajonda MD, Roger L. Hall AAS, Rachele Brassard BSW, Tiffany Bisanar RN, BSN, Mary Cooter MS, Yanne Toulgoat-Dubois BA, Peter Waweru CCRP.
Behavioral Medicine: Michael A. Babyak PhD, James A. Blumenthal PhD, Jeffrey N. Browndyke PhD, Kathleen A. Welsh-Bohmer PhD.
Cardiology: Michael H. Sketch Jr. MD.
Neurology: Ellen R. Bennett PhD, Carmelo Graffagnino MD, Daniel T. Laskowitz MD, Warren J. Strittmatter MD.
Perfusion Services: Kevin Collins BS, CCP, Greg Smigla BS, CCP, Ian Shearer BS, CCP.
Surgery: Thomas A. D’Amico MD, Mani A. Daneshmand MD, R. Jeffrey G. Gaca MD, Donald D. Glower MD, Jack Haney MD, R. David Harpole MD, Mathew G. Hartwig MD, G. Chad Hughes MD, Robert D.B. Jaquiss MD, Shu S. Lin MD, Andrew J. Lodge MD, Carmelo A. Milano MD, Mark W. Onaitis MD, Jacob N. Schroeder MD, Peter K. Smith MD, Betty C. Tong MD.
Rights and permissions
About this article

Cite this article
Klinger, R.Y., Cooter, M., Berger, M. et al. Effect of intravenous lidocaine on the transcerebral inflammatory response during cardiac surgery: a randomized-controlled trial. Can J Anesth/J Can Anesth 63, 1223–1232 (2016). https://doi.org/10.1007/s12630-016-0704-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12630-016-0704-0