
Abstract
Fullerene and its derivatives may bind to biological molecules, causing inhibitory effects. In this context, investigations of interactions of fullerene-based conjugates with proteins are of general interest. Particularly, fullerene and its derivatives demonstrate antibacterial properties; and one of the potential targets for drug design and health therapy is the inhibition of 6-oxopurine phosphoribosyltransferase in Mycobacterium tuberculosis (PDB code: 4RHY). In this article, the binding interactions between a series of quinazoline-4(3H)-ones and their fullerene derivatives with the target transferase were computationally investigated. Initially, we developed predictive quantitative structure-activity relationships (QSAR) models. Next, we introduced a simplified calculation schema that allows to evaluate relative binding affinities and to reveal specific mechanisms of action. For this purpose, the molecular docking approach was utilized to identify the native poses of the 18 transferase inhibitors. The binding pocket of the target protein was isolated and semi-empirical, and hybrid ONIOM scoring functions at different levels of theory were used to treat the ligands and the isolated binding pocket. The agreement within the calculated binding-free energies trends, as well as the agreement with the experimental data, suggests that the developed calculation schema can be used to estimate relative binding affinities towards 4RH. The combination of quantum-chemical models and QSAR models could be applied for future design of new selective inhibitors.





Similar content being viewed by others
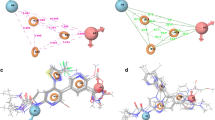

References
WHO media centre (2017) Tuberculosis fact sheet
Smith I (2003) Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev 16:463–496
Hoagland DT, Liu J, Lee RB, Lee RE (2016) New agents for the treatment of drug-resistant mycobacterium tuberculosis. Adv Drug Deliv Rev 102:55–72
Durdagi S, Mavromoustakos T, Chronakis N, Papadopoulos MG (2008) Computational design of novel fullerene analogues as potential HIV-1 PR inhibitors: analysis of the binding interactions between fullerene inhibitors and HIV-1 PR residues using 3D QSAR, molecular docking and molecular dynamics simulations. Bioorg Med Chem 16:9957–9974
Kornev AB, Khakina EA, Troyanov SI, Kushch AA, Peregudov A, Vasilchenko A, Deryabin DG, Martynenko VM, Troshin PA (2012) Facile preparation of amine and amino acid adducts of [60]fullerene using chlorofullerene C60Cl6 as a precursor. Chem Commun 48:5461–5463
Kumar A, Menon SK (2009) Fullerene–ferrocene dyad linked by rigid bilinkage: synthesis, photophysical properties and application as copper ion sensor. J Phys Org Chem 22:661–669
Bobylev AG, Kornev AB, Bobyleva LG, Shpagina MD, Fadeeva IS, Fadeev RS, Deryabin DG, Balzarini J, Troshin PA, Podlubnaya ZA (2011) Fullerenolates: metallated polyhydroxylated fullerenes with potent anti-amyloid activity. Org Biomol Chem 9:5714–5719
Kumar A, Patel B, Patel G, Menon SK (2010) Potentiometric biosensing of glucose by fullerene-based silver selective electrodes. Fullerenes Nanotubes Carbon Nanostructur 18:186–197
Montellano A, Da Ros T, Bianco A, Prato M (2011) Fullerene C60 as a multifunctional system for drug and gene delivery. Nano 3:4035–4041
Foley S, Crowley C, Smaihi M, Bonfils C, Erlanger BF, Seta P, Larroque C (2002) Cellular localisation of a water-soluble fullerene derivative. Biochem Biophys Res Commun 294:116–119
Saleh NA (2015) The QSAR and docking calculations of fullerene derivatives as HIV-1 protease inhibitors. Spectrochim Acta A Mol Biomol Spectrosc 136:1523–1529
Shoji M, Takahashi E, Hatakeyama D, Iwai Y, Morita Y, Shirayama R, Echigo N, Kido H, Nakamura S, Mashino T, Okutani T, Kuzuhara T (2013) Anti-influenza activity of C60 fullerene derivatives. PLoS One 8:e66337
Ahmed L, Rasulev B, Turabekova M, Leszczynska D, Leszczynski J (2013) Receptor- and ligand-based study of fullerene analogues: comprehensive computational approach including quantum-chemical, QSAR and molecular docking simulations. Org Biomol Chem 11:5798–5808
Sizochenko N, Kuz’min V, Ognichenko L, Leszczynski J (2016) Introduction of simplex-informational descriptors for QSPR analysis of fullerene derivatives. J Math Chem 54:698–706
Kar S, Sizochenko N, Ahmed L, Batista VS, Leszczynski J (2016) Quantitative structure-property relationship model leading to virtual screening of fullerene derivatives: exploring structural attributes critical for photoconversion efficiency of polymer solar cell acceptors. Nano Energy 26:677–691
Kuz’min V, Ognichenko L, Mouats A, Artemenko A, Burdina I, Shapkin V, Sizochenko N, Leszczynski J (2016) Efficacy of topological informational potentials for analysis of nonequivalent atoms in molecular graphs: the case of chiral fullerenes. J Math Chem 54:1986–1996
Yilmaz H, Ahmed L, Rasulev B, Leszczynski L (2016) Application of ligand- and receptor-based approaches for prediction of the HIV-RT inhibitory activity of fullerene derivatives. J Nanopart Res 18:123
Ahmed L, Rasulev B, Kar S, Krupa P, Mozolewska MA, Leszczynski J (2017) Inhibitors or toxins? Large library target-specific screening of fullerene-based nanoparticles for drug design purpose. Nano 9:10263–10276
Koul A, Arnoult E, Lounis N, Guillemont J, Andries K (2011) The challenge of new drug discovery for tuberculosis. Nature 469:483–490
Barksdale L, Kim KS (1977) Mycobacterium. Bacteriol Rev 41:217–372
Patel MB, Kumar SP, Valand NN, Jasrai YT, Menon SK (2013) Synthesis and biological evaluation of cationic fullerene quinazolinone conjugates and their binding mode with modeled mycobacterium tuberculosis hypoxanthine-guanine phosphoribosyltransferase enzyme. J Mol Model 19:3201–3217
Tang YJ, Ashcroft JM, Chen D, Min G, Kim CH, Murkhejee B, Larabell C, Keasling JD, Chen FF (2007) Charge-associated effects of fullerene derivatives on microbial structural integrity and central metabolism. Nano Lett 7:754–760
Gastreich M, Lemmen C, Briem H, Rarey M (2005) Addressing the virtual screening challenge, in: virtual screening in drug discovery. CRC Press
Rarey M, Kramer B, Lengauer T, Klebe G (1996) A fast flexible docking method using an incremental construction algorithm. J Mol Biol 261:470–489
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian, Inc., Wallingford
Cossi M, Barone V, Cammi R, Tomasi J (1996) Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem Phys Lett 255:327–335
Sizochenko N, Majumdar D, Roszak S, Leszczynski J (2017) In: Leszczynski J (ed) Handbook of computational chemistry. Springer Netherlands, Dordrecht
Gramatica P, Chirico N, Papa E, Cassani S, Kovarich S (2013) QSARINS: a new software for the development, analysis, and validation of QSAR MLR models. J Comput Chem 34:2121–2132
Puzyn T, Leszczynski J, Cronin MT (2010) Recent advances in QSAR studies: methods and applications. Springer Netherlands, Dordrecht
Roy K, Chakraborty P, Mitra I, Ojha PK, Kar S, Das RN (2013) Some case studies on application of “rm2” metrics for judging quality of quantitative structure–activity relationship predictions: emphasis on scaling of response data. J Comput Chem 34:1071–1082
Tropsha A (2010) Best practices for QSAR model development, validation, and exploitation. Mol Inf 29:476–488
Meng XY, Zhang HX, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 7:146–157
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
Cao Z, Peng Y, Li S, Liu L, Yan T (2008) Molecular dynamics simulation of fullerene C60 in ethanol solution. J Phys Chem C 113:3096–3104
Yang Z, Wang Z, Tian X, Xiu P, Zhou R (2012) Amino acid analogues bind to carbon nanotube via π-π interactions: comparison of molecular mechanical and quantum mechanical calculations. J Chem Phys 136:01B607
Maseras F, Morokuma K (1995) IMOMM: a new integrated ab initio + molecular mechanics geometry optimization scheme of equilibrium structures and transition states. J Comput Chem 16:1170–1179
Dapprich S, Komáromi I, Byun KS, Morokuma K, Frisch MJ (1999) A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J Mol Struct THEOCHEM 461:1–21
Humbel S, Sieber S, Morokuma K (1996) The IMOMO method: integration of different levels of molecular orbital approximations for geometry optimization of large systems: test for n-butane conformation and SN 2 reaction: RCl+ cl−. J Chem Phys 105:1959–1967
Kuno M, Hannongbua S, Morokuma K (2003) Theoretical investigation on nevirapine and HIV-1 reverse transcriptase binding site interaction, based on ONIOM method. Chem Phys Lett 380:456–463
Svensson M, Humbel S, Froese RDJ, Matsubara T, Sieber S, Morokuma K (1996) ONIOM: a multilayered integrated MO+ MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt (P (t-Bu) 3) 2+ H2 oxidative addition. J Phys Chem 100:19357–19363
Vreven T, Mennucci B, da Silva CO, Morokuma K, Tomasi J (2001) The ONIOM-PCM method: combining the hybrid molecular orbital method and the polarizable continuum model for solvation. Application to the geometry and properties of a merocyanine in solution. J Chem Phys 115:62
Cui Q, Guo H, Karplus M (2002) Combining ab initio and density functional theories with semiempirical methods. J Chem Phys 117:5617–5631
Torrent M, Vreven T, Musaev DG, Morokuma K, Farkas Ö, Schlegel HB (2002) Effects of the protein environment on the structure and energetics of active sites of metalloenzymes. ONIOM study of methane monooxygenase and ribonucleotide reductase. J Am Chem Soc 124:192–193
Karelson M, Lobanov VS, Katritzky AR (1996) Quantum-chemical descriptors in QSAR/QSPR studies. Chem Rev 96:1027–1044
Sizochenko N, Gajewicz A, Leszczynski J, Puzyn T (2016) Causation or only correlation? Application of causal inference graphs for evaluating causality in nano-QSAR models. Nano 8:7203–7208
San Diego: Dassault Systèmes (2017) Discovery Studio Modeling Environment
Acknowledgements
The authors thank the NSF-CREST Interdisciplinary Nanotoxicity Center’s NSF-CREST grants # HRD-0833178 and # HRD-1547754 for the support, and want to acknowledge the Extreme Science and Engineering Discovery Environment (XSEDE) by National Science Foundation grant number OCI-1053575 and XSEDE award allocation number DMR110088. The authors thank the Mississippi Center for Supercomputer Research (Oxford, MS) for a generous allotment of computer time. N.S. expresses her gratitude to BioSolveIT GmbH for licensed product. Results obtained using BioSolveIT software are the part of research conducted in the framework of Scientific Challenge fall 2016 organized by BioSolveIT GmbH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 80 kb)
Rights and permissions
About this article

Cite this article
Mirchi, A., Sizochenko, N. & Leszczynski, J. Fullerene quinazolinone conjugates targeting Mycobacterium tuberculosis: a combined molecular docking, QSAR, and ONIOM approach. Struct Chem 29, 765–775 (2018). https://doi.org/10.1007/s11224-018-1100-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-018-1100-x