
Abstract
In this work we have developed a hybrid QM and MM approach to predict pKa of small drug-like molecules in explicit solvent. The gas phase free energy of deprotonation is calculated using the M06-2X density functional theory level with Pople basis sets. The solvation free energy difference of the acid and its conjugate base is calculated at MD level using thermodynamic integration. We applied this method to the 24 drug-like molecules in the SAMPL6 blind pKa prediction challenge. We achieved an overall RMSE of 2.4 pKa units in our prediction. Our results show that further optimization of the protocol needs to be done before this method can be used as an alternative approach to the well established approaches of a full quantum level or empirical pKa prediction methods.




Similar content being viewed by others
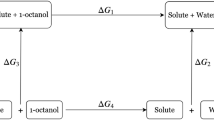
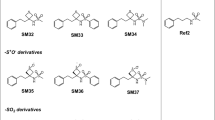
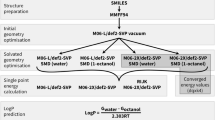
References
Muckerman JT, Skone JH, Ning M, Wasada-Tsutsui Y (2013) Toward the accurate calculation of pKa values in water and acetonitrile. Biochimica et Biophysica Acta (BBA) Bioenergetics 1827(8–9):882–891. https://doi.org/10.1016/j.bbabio.2013.03.011
Seybold PG, Shields GC (2015) Computational estimation of pKa values. Wiley Interdisc Rev: Comput Mol Sci 5(3):290–297. https://doi.org/10.1002/wcms.1218
Wang Y, Xing J, Yuan X, Zhou N, Peng J, Xiong Z, Liu X, Luo X, Luo C, Chen K et al (2015) In silico adme/t modelling for rational drug design. Q Rev Biophys 48(4):488–515. https://doi.org/10.1017/S0033583515000190
Hajjar E, Dejaegere A, Reuter N (2009) Challenges in pKa predictions for proteins: the case of asp213 in human proteinase 3. J Phys Chem A 113(43):11783–11792. https://doi.org/10.1021/jp902930u
Lee AC, Crippen GM (2009) Predicting pKa. J Chem Inf Model 49(9):2013–2033. https://doi.org/10.1021/ci900209w
Zevatskii YE, Samoilov DV (2011) Modern methods for estimation of ionization constants of organic compounds in solution. Russ J Org Chem 47(10):1445–1467. https://doi.org/10.1134/s1070428011100010
Greenwood JR, Calkins D, Sullivan AP, Shelley JC (2010) Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J Comput-Aided Mol Des 24(6–7):591–604. https://doi.org/10.1007/s10822-010-9349-1
Fraczkiewicz R, Lobell M, Gller AH, Krenz U, Schoenneis R, Clark RD, Hillisch A (2014) Best of both worlds: combining pharma data and state of the art modeling technology to improve in silico pKa prediction. J Chem Inf Model 55(2):389–397. https://doi.org/10.1021/ci500585w
Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M (2007) Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J Comput-Aided Mol Des 21(12):681–691. https://doi.org/10.1007/s10822-007-9133-z
Li M, Zhang H, Chen B, Wu Y, Guan L (2018) Prediction of pKa values for neutral and basic drugs based on hybrid artificial intelligence methods. Sci Rep 8(1):3991. https://doi.org/10.1038/s41598-018-22332-7
Bochevarov AD, Watson MA, Greenwood JR, Philipp DM (2016) Multiconformation, density functional theory-based pKa prediction in application to large, flexible organic molecules with diverse functional groups. J Chem Theory Comput 12(12):6001–6019. https://doi.org/10.1021/acs.jctc.6b00805
Klamt A, Eckert F, Diedenhofen M, Beck ME (2003) First principles calculations of aqueous pKa values for organic and inorganic acids using cosmors reveal an inconsistency in the slope of the pKa scale. J Phys Chem A 107(44):9380–9386. https://doi.org/10.1021/jp034688o
Klici JJ, Friesner RA, Liu S-Y, Guida WC (2002) Accurate prediction of acidity constants in aqueous solution via density functional theory and self-consistent reaction field methods. J Phys Chem A 106(7):1327–1335. https://doi.org/10.1021/jp012533f
Thapa B, Bernhard Schlegel H (2017) Improved pKa prediction of substituted alcohols, phenols, and hydroperoxides in aqueous medium using density functional theory and a cluster-continuum solvation model. J Phys Chem A 121(24):4698–4706. https://doi.org/10.1021/acs.jpca.7b03907
Ho J (2015) Are thermodynamic cycles necessary for continuum solvent calculation of pKas and reduction potentials? Phys Chem Chem Phys 17(4):2859–2868. https://doi.org/10.1039/c4cp04538f
Lian P, Johnston RC, Parks JM, Smith JC (2018) Quantum chemical calculation of pKas of environmentally relevant functional groups: carboxylic acids, amines, and thiols in aqueous solution. J Phys Chem A 122(17):4366–4374. https://doi.org/10.1021/acs.jpca.8b01751
Riojas AG, Wilson AK (2014) Solv-ccca: implicit solvation and the correlation consistent composite approach for the determination of pKa. J Chem Theory Comput 10(4):1500–1510. https://doi.org/10.1021/ct400908z
Liptak MD, Shields GC (2001) Accurate pKa calculations for carboxylic acids using complete basis set and gaussian-n models combined with cpcm continuum solvation methods. J Am Chem Soc 123(30):7314–7319. https://doi.org/10.1021/ja010534f
Liptak MD, Shields GC (2001) Experimentation with different thermodynamic cycles used for pKa calculations on carboxylic acids using complete basis set and gaussian-n models combined with cpcm continuum solvation methods. Int J Quantum Chem 85(6):727–741. https://doi.org/10.1002/qua.1703
Tehan BG, Lloyd EJ, Wong MG, Pitt WR, Montana JG, Manallack DT, Gancia E (2002) Estimation of pKa using semiempirical molecular orbital methods. part 1: application to phenols and carboxylic acids. Quant Struct-Act Relat 21(5):457–472. https://doi.org/10.1002/1521-3838(200211)21:5<457::aid-qsar457>3.0.co;2-5.
Peverati R, Truhlar DG (2014) Quest for a universal density functional: the accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos Trans R Soc A 372(2011):20120476–20120476. https://doi.org/10.1098/rsta.2012.0476
Klamt A, Schrmann G (1993) Cosmo: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans 2(5):799–805. https://doi.org/10.1039/p29930000799
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396. https://doi.org/10.1021/jp810292n
Ho J, Ertem MZ (2016) Calculating free energy changes in continuum solvation models. J Phys Chem B 120(7):1319–1329. https://doi.org/10.1021/acs.jpcb.6b00164
Barone V, Cossi M (1998) Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A 102(11):1995–2001. https://doi.org/10.1021/jp9716997
Ho J (2014) Predicting pKa in implicit solvents: current status and future directions. Aust J Chem 67(10):1441. https://doi.org/10.1071/ch14040
Casasnovas R, Ortega-Castro J, Frau J, Donoso J, Muoz F (2014) Theoretical pKa calculations with continuum model solvents, alternative protocols to thermodynamic cycles. Int J Quantum Chem 114(20):1350–1363. https://doi.org/10.1002/qua.24699
Muddana HS, Sapra NV, Fenley AT, Gilson MK (2014) The SAMPL4 hydration challenge: evaluation of partial charge sets with explicit-water molecular dynamics simulations. J Comput-Aided Mol Des 28(3):277–287. https://doi.org/10.1007/s10822-014-9714-6
König G, Pickard FC, Mei Y, Brooks BR (2014) Predicting hydration free energies with a hybrid qm/mm approach: an evaluation of implicit and explicit solvation models in SAMPL4. J Comput-Aided Mol Des 28(3):245–257. https://doi.org/10.1007/s10822-014-9708-4
Mobley DL, Bayly CI, Cooper MD, Shirts MR, Dill KA (2015) Correction to small molecule hydration free energies in explicit solvent: an extensive test of fixed-charge atomistic simulations. J Chem Theory Comput 11(3):1347–1347. https://doi.org/10.1021/acs.jctc.5b00154
Shirts MR, Pitera JW, Swope WC, Pande VS (2003) Extremely precise free energy calculations of amino acid side chain analogs: comparison of common molecular mechanics force fields for proteins. J Chem Phys 119(11):5740–5761. https://doi.org/10.1063/1.1587119
Peter J (2009) Guthrie. A blind challenge for computational solvation free energies: introduction and overview. J Phys Chem B 113(14):4501–4507. https://doi.org/10.1021/jp806724u
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) The SAMPL4 hostguest blind prediction challenge: an overview. J Comput-Aided Mol Des 28(4):305–317. https://doi.org/10.1007/s10822-014-9735-1
Pickard FC, Knig G, Tofoleanu F, Lee J, Simmonett AC, Shao Y, Ponder JW, Brooks BR (2016) Blind prediction of distribution in the sampl5 challenge with qm based protomer and pKa corrections. J Comput-Aided Mol Des 30(11):1087–1100. https://doi.org/10.1007/s10822-016-9955-7
Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL, Gilson MK (2016) Overview of the sampl5 hostguest challenge: are we doing better? J Comput-Aided Mol Des 31(1):1–19. https://doi.org/10.1007/s10822-016-9974-4
Geballe MT, Guthrie JP (2012) The sampl3 blind prediction challenge: transfer energy overview. J Comput-Aided Mol Des 26(5):489–496. https://doi.org/10.1007/s10822-012-9568-8
Rustenburg AS, Dancer J, Lin B, Feng JA, Ortwine DF, Mobley DL, Chodera JD (2016) Measuring experimental cyclohexane-water distribution coefficients for the sampl5 challenge. J Comput-Aided Mol Des 30(11):945–958. https://doi.org/10.1007/s10822-016-9971-7
Isik M (2018) pKa measurements for the sampl6 prediction challenge for a set of kinase inhibitor-like fragments. J Comput-Aided Mol Des. https://doi.org/10.1101/368787
Szakács Z, Noszál B (1999) Protonation microequilibrium treatment of polybasic compounds with any possible symmetry. J Math Chem 26(1):139
Philipp DM, Watson MA, Yu HS, Steinbrecher TB, Bochevarov AD (2018) Quantum chemical prediction for complex organic mole. Int J Quantum Chem 118(12):e25561. https://doi.org/10.1002/qua.25561
Darvey IG (1995) The assignment of pKa values to functional groups in amino acids. Biochem Educ 23(2):80–82. https://doi.org/10.1016/0307-4412(94)00150-n
McQuarrie DA (2000) Statistical mechanics. University Science Books, Sausalito
Tissandier MD, Cowen KA, Feng WY, Gundlach E, Cohen MH, Earhart AD, Coe JV, Tuttle TR (1998) The proton’s absolute aqueous enthalpy and gibbs free energy of solvation from cluster-ion solvation data. J Phys Chem A 102(40):7787–7794. https://doi.org/10.1021/jp982638r
Jorgensen WL, Ravimohan C (1985) Monte carlo simulation of differences in free energies of hydration. J Chem Phys 83(6):3050–3054. https://doi.org/10.1063/1.449208
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian16 Revision B.01. GaussianInc., Wallingford, CT
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) Charmm: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4(2):187–217. https://doi.org/10.1002/jcc.540040211
Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S et al (2009) Charmm: the biomolecular simulation program. J Comput Chem 30(10):1545–1614. https://doi.org/10.1002/jcc.21287
Anderson Eric, Veith GD, Weininger D (1987) SMILES, a line notation and computerized interpreter for chemical structures. U.S. Environmental Protection Agency, Environmental Research Laboratory, Duluth
Mazzatorta P, Tran L-A, Schilter B, Grigorov M (2007) Integration of structure activity relationship and artificial intelligence systems to improve in silico prediction of ames test mutagenicity. ChemInform. https://doi.org/10.1002/chin.200715211
Zhao Y, Truhlar DG (2007) The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four m06-class functionals and 12 other functionals. Theor Chem Acc 120(1–3):215–241. https://doi.org/10.1007/s00214-007-0310-x
MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiskiewicz-Kuczera J, Yin D, Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102(18):3586–3616. https://doi.org/10.1021/jp973084f
Jakalian A, Jack DB, Bayly CI (2002) Fast, efficient generation of high-quality atomic charges. am1-bcc model: II. Parameterization and validation. J Comput Chem 23(16):1623–1641. https://doi.org/10.1002/jcc.10128
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25(9):1157–1174. https://doi.org/10.1002/jcc.20035
Evans DJ, Holian BL (1985) The nosehoover thermostat. J Chem Phy 83(8):4069–4074. https://doi.org/10.1063/1.449071
Darden T, York D, Pedersen L (1993) Particle mesh ewald: an nlog(n) method for ewald sums in large systems. J Chem Phys 98(12):10089–10092. https://doi.org/10.1063/1.464397
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh ewald method. J Chem Phys 103(19):8577–8593. https://doi.org/10.1063/1.470117
Kuhn HW (1955) The hungarian method for the assignment problem. Naval Res Logist Q 2(12):83–97. https://doi.org/10.1002/nav.3800020109
Vanommeslaeghe K, Mackerell AD (2012) Automation of the charmm general force field (cgenff) I: bond perception and atom typing. J Chem Inf Model 52(12):3144–3154. https://doi.org/10.1021/ci300363c
Mayne CG, Gumbart JC, Tajkhorshid E (2013) The force field toolkit: software for the parameterization of small molecules from first principles. Biophys J 104(2):31a. https://doi.org/10.1016/j.bpj.2012.11.209
Huang L, Roux B (2013) Automated force field parameterization for nonpolarizable and polarizable atomic models based on ab initio target data. J Chem Theory Comput 9(8):3543–3556. https://doi.org/10.1021/ct4003477
Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the gromos force-field parameter sets 53a5 and 53a6. J Comput Chem 25(13):1656–1676. https://doi.org/10.1002/jcc.20090
Miguel ELM, Santos CIL, Silva CM, Pliego JR Jr (2016) How accurate is the SMD model for predicting free energy barriers for nucleophilic substitution reactions in polar protic and dipolar aprotic solvents? J Braz Chem Soc 27:2055–2061. https://doi.org/10.5935/0103-5053.20160095
Lee J, Miller BT, Brooks BR (2015) Computational scheme for pH-dependent binding free energy calculation with explicit solvent. Protein Sci 25(1):231–243. https://doi.org/10.1002/pro.2755
Knig G, Brooks BR (2015) Correcting for the free energy costs of bond or angle constraints in molecular dynamics simulations. Biochim Biophys Acta (BBA) 1850(5):932–943. https://doi.org/10.1016/j.bbagen.2014.09.001
Khandogin J, Brooks CL (2005) Constant pH molecular dynamics with proton tautomerism. Biophys J 89:141–157
Donnini S, Tegeler F, Groenhof G, Grubmuller H (2011) Constant pH molecular dynamics in explicit solvent with \(\lambda\)-dynamics. J Chem Theory Comput 7:1962–1978
Tao P, Sodt AJ, Shao Y, Knig G, Brooks BR (2014) Computing the free energy along a reaction coordinate using rigid body dynamics. J Chem Theory Comput 10(10):4198–4207. https://doi.org/10.1021/ct500342h
Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Schnieders MJ, Haque I, Mobley DL, Lambrecht DS, DiStasio RA, Head-Gordon M, Clark GNI, Johnson ME, Head-Gordon T (2010) Current status of the amoeba polarizable force field. J Phys Chem B 114(8):2549–2564
Bradshaw RT, Essex JW (2016) Evaluating parametrization protocols for hydration free energy calculations with the amoeba polarizable force field. J Chem Theory Comput 12(8):3871–3883. https://doi.org/10.1021/acs.jctc.6b00276
Baker CM, Lopes PEM, Zhu X, Benoit R, Mackerell AD (2010) Accurate calculation of hydration free energies using pair-specific lennard-jones parameters in the charmm drude polarizable force field. J Chem Theory Comput 6(4):1181–1198
Huang J, Simmonett AC, Pickard FC, Mackerell AD, Brooks BR (2017) Mapping the drude polarizable force field onto a multipole and induced dipole model. J Chem Phys 147(16):161702. https://doi.org/10.1063/1.4984113
Acknowledgements
The work is supported by the Intramural Research Program of the National Heart, Lung and Blood Institute Z01 HL001051. The authors would like to acknowledge Xiongwu Wu, Kyungreem Han, Philip Hudson, Michael Jones, Ana Damjanovic, Gerhard Konig, Frank Pickard, Florentina Tofoleanu, Reuben Meanapa for helpful discussion. This work utilized the computational resources of the NIH HPC Biowulf cluster. http://hpc.nih.gov and the Laboratory of Computational Biology cluster. SP would like to acknowledge Biochemistry, Cellular and Molecular Biology (BCMB) graduate program at JHMI.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Prasad, S., Huang, J., Zeng, Q. et al. An explicit-solvent hybrid QM and MM approach for predicting pKa of small molecules in SAMPL6 challenge. J Comput Aided Mol Des 32, 1191–1201 (2018). https://doi.org/10.1007/s10822-018-0167-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-018-0167-1