
Abstract
The nitrogen and methane cycles are important biogeochemical processes. Recently, ‘Candidatus Methanoperedens nitroreducens,’ archaea that catalyze nitrate-dependent anaerobic oxidation of methane (AOM), were enriched, and their genomes were analyzed. Diagnostic molecular tools for the sensitive detection of ‘Candidatus M. nitroreducens’ are not yet available. Here, we report the design of two novel mcrA primer combinations that specifically target the alpha sub-unit of the methyl-coenzyme M reductase (mcrA) gene of ‘Candidatus M. nitroreducens’. The first primer pair produces a fragment of 186-bp that can be used to quantify ‘Candidatus M. nitroreducens’ cells, whereas the second primer pair yields an 1191-bp amplicon that is with sufficient length and well suited for more detailed phylogenetic analyses. Six different environmental samples were evaluated with the new qPCR primer pair, and the abundances were compared with those determined using primers for the 16S rRNA gene. The qPCR results indicated that the number of copies of the ‘Candidatus M. nitroreducens’ mcrA gene was highest in rice field soil, with 5.6 ± 0.8 × 106 copies g−1 wet weight, whereas Indonesian river sediment had only 4.6 ± 2.7 × 102 copies g−1 wet weight. In addition to freshwater environments, sequences were also detected in marine sediment of the North Sea, which contained approximately 2.5 ± 0.7 × 104 copies g−1 wet weight. Phylogenetic analysis revealed that the amplified 1191-bp mcrA gene sequences from the different environments all clustered together with available genome sequences of mcrA from known ‘Candidatus M. nitroreducens’ archaea. Taken together, these results demonstrate the validity and utility of the new primers for the quantitative and sensitive detection of the mcrA gene sequences of these important nitrate-dependent AOM archaea. Furthermore, the newly obtained mcrA sequences will contribute to greater phylogenetic resolution of ‘Candidatus M. nitroreducens’ sequences, which have been only poorly captured by general methanogenic mcrA primers.
Similar content being viewed by others


Introduction
Methane is an important greenhouse gas (GHG) that contributes approximately 20% to global warming (Myhre et al. 2013). Since the advent of industrialization, atmospheric concentrations of methane have increased by 150%, potentially further exacerbating climate change (Schwietzke et al. 2016). Evaluating the contribution of environmental microorganisms that produce or consume this significant GHG is essential for understanding methane sources and sinks and developing mitigation strategies for methane released into the atmosphere. Most research on microorganisms involved in the methane cycle has focused on aerobic methanotrophic bacteria that inhabit oxic environments or archaea that produce methane in anoxic zones. However, recent studies have revealed that in the anoxic layers of soils and sediments, methane is consumed by anaerobic methanotrophic bacteria and/or archaea that use alternate electron acceptors such as nitrite, nitrate, or iron (Egger et al. 2015; Ettwig et al. 2010; Raghoebarsing et al. 2006).
Enrichment cultures inoculated with freshwater sediment exhibited coupling of the reduction of nitrite to the anaerobic oxidation of methane (Ettwig et al. 2008; Raghoebarsing et al. 2006). The corresponding nitrite-dependent methanotrophic bacteria were identified as belonging to the bacterial NC10 phylum and named ‘Candidatus Methylomirabilis oxyfera’ (Ettwig et al. 2010). This microorganism exhibits an intra-aerobic metabolism in which nitric oxide is hypothesized to be dismutated to oxygen and nitrogen gas. The oxygen could subsequently be used by the canonical particulate methane monooxygenase encoded by pmoCAB.
Archaea that oxidize methane anaerobically were initially discovered in marine environments, where they carry out sulfate-dependent anaerobic oxidation of methane (S-AOM). These anaerobic methane-oxidizing archaea (ANME) have been estimated to oxidize up to 90% of released methane before it reaches the atmosphere (Hinrichs and Boetius 2002; Knittel and Boetius 2009). ANMEs are divided into three lineages, ANME-1, ANME-2, and ANME-3 (Knittel et al. 2005; Nauhaus et al. 2005; Stadnitskaia et al. 2005) and are further divided into sub-clades in some cases. All three lineages have been detected in marine and freshwater environments.
Recently, the genomes of ANME-2d archaea enriched in bioreactors fed with methane, nitrate, and ammonium or methane and nitrate were obtained (Arshad et al. 2015; Haroon et al. 2013). These Euryarchaea, which are capable of coupling nitrate reduction to anaerobic methane oxidation, were identified as ‘Candidatus Methanoperedens nitroreducens.’ Phylogenetic analysis revealed that these archaea are related to Methanosarcina in the Methanosarcinales order (Haroon et al. 2013) and are classified as GOM Arc I in the ribosomal RNA (rRNA) SILVA database. The GOM Arc I consists of the ANME-2d group as well as the original GOM Arc I group with sequences from the Gulf of Mexico (Mills et al. 2003).
‘Candidatus M. nitroreducens’ possesses all genes of the (reverse) methanogenic pathway (Arshad et al. 2015; Haroon et al. 2013). The best-characterized enzyme of methanogenesis and AOM is methyl-coenzyme M reductase (MCR). In methanogenesis, MCR catalyzes the terminal step of the pathway, resulting in the release of methane. In the anaerobic oxidation of methane, MCR functions in a reverse mode (Hallam et al. 2003, 2004; Krüger et al. 2003), catalyzing the activation of methane (Krüger et al. 2003). The genomes of two ‘Candidatus M. nitroreducens’ strains have been assembled and analyzed. In both genome assemblies, the complete reverse methanogenesis pathway including the mcrABCDG genes was identified (Arshad et al. 2015; Haroon et al. 2013), and the genomes contained only a single copy of the 16S rRNA and the mcrA gene. Furthermore, the enzymes for nitrate reduction to nitrite and nitrite reduction to ammonium appeared to be encoded by narGH- and nrf-type genes, respectively (Arshad et al. 2015).
For ‘Candidatus M. oxyfera’ bacteria, specific primers for both the 16S rRNA gene and the pmoA gene have been designed (Ettwig et al. 2009; Luesken et al. 2011). Analyses of various environmental samples using these primers have demonstrated that ‘Candidatus M. oxyfera’ is present in peat lands, lake sediments, wastewater treatment systems, rice fields, and various other anoxic environments (Deutzmann and Schink 2011; Hu et al. 2014; Zhou et al. 2014; Zhu et al. 2012). As nitrate concentrations in freshwater environments are generally higher than those of nitrite or sulfate, ‘Candidatus M. nitroreducens’ may contribute significantly to nitrate-dependent AOM in these environments (Vaksmaa et al. 2016). To detect ‘Candidatus M. nitroreducens’ in environmental samples, specific fluorescence in situ hybridization (FISH) probes have been designed (Schubert et al. 2011). The development of quantitative detection methods based on the 16S rRNA gene has also been reported (Ding et al. 2015).
Although the 16S rRNA gene is most commonly used for phylogenetic surveys, the mcrA gene is an alternative and more specific biomarker for the detection of methanogens and ANMEs in the environment. Although previously published mcrA primers were designed to mainly target all known methanogens and ANMEs, most have a strong bias toward certain methanogens or specific groups of ANMEs (Hales et al. 1996; Juottonen et al. 2006; Luton et al. 2002; Nunoura et al. 2008). Available general mcrA primers are not well suited to capturing mcrA sequences of ‘Candidatus M. nitroreducens’ in the environment, potentially resulting in underrepresentation in molecular surveys. Furthermore, differentiating between phylogenetically closely related methanogens and methanotrophs is crucial to directly link observed diversity with the organisms responsible for either methane oxidation or methane production. In the current study, we developed two novel mcrA primer pairs that specifically target ‘Candidatus M. nitroreducens’ for use in quantification and more refined phylogenetic analysis. We used these primers to study the distribution and abundance of ‘Candidatus M. nitroreducens’ in various ecosystems. For comparison, we validated the use of 16S rRNA gene probes designed for FISH analysis as qPCR primers and compared the results with the diversity and abundance obtained with the novel mcrA primers.
Materials and methods
Environmental samples
Environmental samples were obtained from six different locations: rice field soils (Vercelli, Italy), sludge from a brewery wastewater treatment plant (Lieshout, The Netherlands), North Sea sediment (The Netherlands), polluted Citarum River sediment (Indonesia), Jordan River sediment (UT, USA), and State Channel sediment (UT, USA). In addition to the environmental samples, an enrichment culture (AOM enrichment Vercelli) of ‘Candidatus M. nitroreducens’ was used as a sample for primer validation (Vaksmaa et al in preparation). The samples were stored at −20 °C prior to DNA extraction. Detailed information on the geographic locations is presented in Table S1.
Primer design, DNA extraction, and PCR amplification
For primer design, 20,000 high-quality mcrA sequences deposited in the NCBI GenBank database (Benson et al. 2013) were downloaded and aligned, and the lengths of these sequences were inspected. From the alignment of 20,000 mcrA sequences, 45 available full-length mcrA sequences (two belonging to ‘Candidatus Methanoperedens nitroreducens’) were used for primer design using the probe design tool implemented in ARB (Ludwig et al. 2004). The designed mcrA primer set McrA159F/McrA345R amplifies a 186-bp fragment and has a predicted annealing temperature of 62 °C. The McrA169F/McrA1360R primer pair yields a 1191-bp fragment. Detailed information on the mcrA primers and 16S rRNA primers used in this study is provided in Table 1. Commonly used general mcrA gene primers were in silico evaluated for their ability to target ‘Candidatus Methanoperedens nitroreducens,’ and the number of mismatches is brought out in Table 2. For comparison, the 16S rRNA gene of ‘Candidatus Methanoperedens nitroreducens’ was targeted with the clade-specific primers AAA641F and AAA834R (previously reported as FISH probes) (Schubert et al. 2011). These primers amplify a 212-bp fragment with an optimal annealing temperature of 60 °C. DNA was extracted from all samples with the PowerSoil® DNA Isolation Kit. First, 0.1–0.35 g of soil was weighed into the 2-ml tubes provided with the kit, which contained buffer and beads. The following steps were performed according to the manufacturer’s protocol (MO BIO Laboratories Inc., Carlsbad, USA). DNA quantity was assessed using a microspectrophotometer (NanoDrop, ND-1000, Isogen Life Science, The Netherlands). All PCR reactions were performed using PerfeCTa Quanta master mix (Quanta Biosciences, Gaithersburg, USA) with the following composition: 1 μl each of 20 μM of the forward and reverse primers, 12.5 μl of PCR master mix and 9.5 μl of Milli-Q water. The PCR temperature gradient program was 96 °C for 5 min, followed by 45 cycles of 96 °C for 30 s, gradient (55–68 °C) for 45 s, and 72 °C for 45 s and a final extension at 72 °C for 10 min.
Cloning, sequencing, and phylogenetic analysis
The sizes of the PCR products obtained with the McrA159F/McrA345R, McrA169F/McrA1360R, or AAA641F/AAA834R primer pairs were evaluated by gel electrophoresis on 1% agarose gels. The fragments were purified using the GeneJET PCR purification kit according to the manufacturer’s protocol (Thermo Scientific, Landsmeer, The Netherlands). The amplified PCR products were cloned using the pGEM-T Easy cloning vector (Promega, USA) and used to transform E. coli XL1 Blue competent cells. The cells were plated on Luria-Bertani (LB) agar plates containing 20 μl of 100 mg/ml ampicillin, 35 μl of 2% X-Gal, and 35 μl of 100 mM IPTG. The plates were incubated at 37 °C overnight. Colony PCR was performed by direct PCR using the M13F and M13R primers. The PCR program consisted of initialization at 96 °C for 10 min, followed by 40 cycles of amplification at 96 °C for 45 s, 57 °C for 30 s, and 72 °C for 30 s and a final elongation step at 72 °C for 5 min. The colonies resulting in amplification of a fragment of the correct size were grown in 5 ml of LB medium overnight at 37 °C prior to plasmid isolation with a GeneJET Plasmid Miniprep Kit (Thermo Scientific, The Netherlands). The inserts were sequenced at BaseClear B.V. (Leiden, Netherlands) or Macrogen (Amsterdam, Netherlands). For short fragments, the MF primer (5′TTTCCCAGTCACGACGTTG′3) was used, and to retrieve longer fragments, sequencing was also performed with the MR primer (5′GGATAACAATTTCACACAGG′3). The quality of the sequences was assessed with the Chromas Lite 2.01 (Technelysium Pty Ltd., Australia) software. All DNA sequences were imported into the mcrA ARB database. ARB version 5.5 was used for phylogenetic comparison (Ludwig et al. 2004). Phylogenetic trees based on the DNA sequences were calculated using the neighbor-joining algorithm with the Jukes-Cantor correction. Sequences were further analyzed by BLASTn and BLASTx at NCBI (Altschul et al. 1990).
Quantification by qPCR
The mcrA and 16S rRNA gene copy numbers in the environmental samples were quantified with the primer set McrA159F/McrA345R and the 16S rRNA gene primers AAA641F/AAA834R. All qPCR reactions were performed using PerfeCTa Quanta master mix (Quanta Biosciences, Gaithersburg, USA) and 96-well optical plates (Bio-Rad Laboratories, Hercules, England). Each reaction was performed in triplicate on duplicate DNA extractions. All reactions were performed using the Bio-Rad IQ™ 5 cycler (Biorad, USA). Negative controls were added to each plate by replacing the sample volume with autoclaved Milli-Q water. Standard curves were constructed by tenfold serial dilution of a known copy number of the pGEM-T easy plasmid with inserted DNA of the target gene.
In silico evaluation of 16S rRNA primers
The specificity and intra-group coverage of the 16S rRNA gene primers DP397F/DP569R (Ding et al. 2015) and the primers AAA641F/AAA834R, which target ‘Candidatus M. nitroreducens,’ were evaluated. The comparison was carried out in ARB (Ludwig et al. 2004) using the GOM Arc I group as a representative group for ‘Candidatus M. nitroreducens’ and related sequences. The specificity and intra-group coverage of both primer sets were evaluated using the non-redundant version of the SILVA SSU Ref dataset (release 119; (Quast et al. 2013)), which contains 535,004 high-quality 16S rRNA gene sequences, of which 109 belong to GOM Arc I.
Nucleotide sequence accession numbers
Representative sequences were deposited at GenBank under the accession numbers KX290067–KX290105 for mcrA sequences amplified with the McrA159F/McrA345R primers and under the accession numbers KX290017–KX290044 for mcrA sequences obtained with the primers McrA169F/McrA1360R. The 16S rRNA gene sequences were deposited under accession numbers KX290045–KX290065.
Results
Specificity of the novel mcrA primers for qPCR
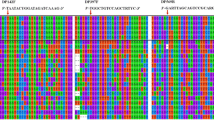
To design an mcrA primer set specific for ‘Candidatus M. nitroreducens,’ available full-length mcrA sequences (45 sequences) covering the diversity of known methanotrophs and methanogens were aligned and used for primer design. Representative sequences and the primer-binding positions are depicted in Fig. 1. Two sites at an appropriate distance for qPCR amplification (at nucleotide positions 159–181 and 322–345, respectively) were conserved between the two ‘Candidatus M. nitroreducens’ sequences but were different in all other archaeal mcrA sequences. The resulting McrA159F/McrA345R primer pair amplifies a fragment of 186 bp, suitable for qPCR. The forward primer McrA159F possesses four mismatches with the mcrA sequence of the closest methanogen, Methanobacterium alcaliphilum. The reverse primer McrA345R possesses three mismatches with the mcrA sequences of the methanogens Methanothermobacter wolfeii and Methanohalophilus halophilus. The optimal annealing temperature of 62 °C was determined by gradient PCR using DNA extracted from rice field soil. All 40 PCR products amplified from DNA from the environmental samples and the enrichment culture were cloned and sequenced and corresponded to the expected part of the mcrA gene. The sequencing resulted in five to seven clone sequences per each environmental sample. All of the sequences had very high similarity to the mcrA gene of the two described ‘Candidatus M. nitroreducens’ strains (91–100% at the nucleotide level and 97–100% at the amino acid level (Table S2)).

Excerpt of the full-length sequence alignment of the mcrA genes of anaerobic methanotrophs and methanogens. The binding sites and conserved positions of the McrA159F forward and McrA345R reverse primers are indicated in 2
qPCR quantification of ‘Candidatus Methanoperedens nitroreducens’ mcrA and 16S rRNA gene copies in environmental samples
The newly designed mcrA primers McrA159F/McrA345R were used with DNA extracted from six environmental samples. In addition, the results were compared with the copy numbers obtained with the primers AAA641F/AAA834R targeting the 16S rRNA gene of ‘Candidatus M. nitroreducens.’
Two 16S rRNA primer sets, the primer pairs DP397F/DP569R (Ding et al. 2015) and AAA641F/AAA834R, have been proposed to target ‘Candidatus M. nitroreducens’ and the GOM Arc I group, respectively. Here, we analyzed the applicability of these primer sets in silico as specific qPCR primers to target the GOM Arc I group. The intra-group coverage and the number of out-group targets with one to three allowed mismatches are presented in Table 3. The primer pair AAA641F/AAA834R exhibited higher intra-group coverage (65–84%) than the DP397F/DP569R primers, which covered less than 60% of the GOM Arc I sequences at zero mismatch. Thus, we experimentally tested the AAA641F/AAA834R primers using DNA from the environmental samples and the enrichment culture and sequenced the PCR products. Twenty-one of the resultant clone sequences were highly similar to the 16S rRNA gene sequences of the two described ‘Candidatus M. nitroreducens’ strains, whereas two clone sequences did not correspond to ‘Candidatus M. nitroreducens’ (Table S3).
In the qPCR analysis, the highest ‘Candidatus M. nitroreducens’ copy numbers were obtained in rice field soil, with an average mcrA gene copy number of 5.6 ± 0.8 × 106 copies g−1 wet weight and an average 16S rRNA gene abundance of 1.3 ± 0.3 × 108 copies g−1 wet weight. Rice field soil was followed by river sediment (State Channel, USA; 4.4 ± 4.4 × 105 mcrA gene copies g−1 wet weight and 1.8 ± 0.6 × 107 16S rRNA gene copies g−1 wet weight), wastewater treatment plant sludge (1.2 ± 0.8 × 105 mcrA gene copies g−1 wet weight and 6.7 ± 2.2 × 107 16S rRNA gene copies g−1 wet weight), Indonesian river sediment (3.0 ± 0.7 × 104 mcrA gene copies g−1 wet weight and 4.2 ± 2.2 × 106 16S rRNA gene copies g−1 wet weight), and North Sea sediment (2.5 ± 0.7 × 104 mcrA gene copies g−1 wet weight and 4.5 ± 0.3 × 106 16S rRNA gene copies g−1 wet weight). The lowest abundance was recorded in the sediment of the Jordan River (UT, USA), where qPCR did not result in any 16S rRNA gene amplification and only 4.6 ± 2.7 × 102 copies of the mcrA gene g−1 wet weight were detected (Fig. 2).

Boxplot depicting the abundance of ‘Candidatus M. nitroreducens’ in environmental samples as assessed by quantitative PCR of the 16S rRNA gene and mcrA gene. For each sample, six independent qPCR reactions of two DNA samples were performed. The environmental samples originated from rice field soil (RF), wastewater treatment plant sludge (BS), North Sea sediment (NS), State Channel sediment (SC), Indonesian river sediment (IR), and Jordan River sediment (JR). The horizontal line within each box represents the median, and the error bars represent the standard deviation. The upper and lower in each box lines represent the 75 and 25 percentiles, respectively. For the Jordan River sediment, no amplification was detected with Methanoperedens-specific 16S rRNA gene primers
Phylogenetic analysis
In addition to the qPCR primers McrA159F/McrA345R, a second primer set was designed to amplify longer mcrA fragments. Conserved regions were identified at nucleotide positions 169–192 and 1336–1360. The resulting primer set, McrA169F/McrA1360R, amplifies a fragment of 1191 bp, suitable for detailed phylogenetic analysis. The primers were again tested using DNA extracted from the environmental samples and the enrichment culture as described in the “Materials and methods” section. Amplification resulted in a single band of the expected size, and sequence analysis indicated that all 40 sequences were highly similar to ‘Candidatus M. nitroreducens.’ The phylogenetic positions of these clones are depicted in Fig. 3. Clustering of sequences from the same environment was not observed, although all sequences clustered more closely with ‘Candidatus Methanoperedens sp. DS-2015’ than ‘Candidatus Methanoperedens nitroreducens ANME-2d.’ On average, the sequences exhibited higher identity to ‘Candidatus Methanoperedens sp. DS-2015’ (87–99% nucleotide sequence identity) than to ‘Candidatus Methanoperedens nitroreducens ANME-2d’ (85–90% nucleotide sequence identity). The sequence identities of all clones to the two described strains are provided in Table S4.

a Phylogenetic overview of methanogenic and anaerobic methanotrophic archaea based on mcrA gene sequences. The phylogenetic position of GOM Arc I archaea is marked in pink. b Phylogenetic tree of ‘Candidatus M. nitroreducens’ mcrA clone sequences (n = 28, 1191 bp). The tree includes the clones derived from this study as well as reference sequences of ‘ Candidatus Methanoperedens nitroreducens ANME-2d’ (GenBank accession number JMY01000002.1) and ‘Candidatus Methanoperedens sp. DS-2015’ (GenBank accession number LKCM01000080.1). The tree was computed using the neighbor-joining algorithm with the Jukes-Cantor correction (Color figure online)
Discussion
In this study, we developed specific and sensitive molecular detection tools to target nitrate-dependent anaerobic methanotrophic ‘Candidatus M. nitroreducens’ archaea. We designed two novel PCR primer sets for the mcrA gene of ‘Candidatus M. nitroreducens,’ thus providing a straightforward detection and quantification method. The primer set McrA159F/McrA345R results in the amplification of a 186-bp fragment and is suitable for quantification of mcrA gene copies by qPCR. The other primer set, McrA169F/McrA1360R, results in the amplification of a 1191-bp fragment that can be used in more accurate and detailed phylogenetic analyses.
The genomes of known ‘Candidatus M. nitroreducens’ strains possess only a single copy of the 16S rRNA gene and the mcrA gene, although copy numbers might differ for non-cultivated species. However, the copy numbers in the environmental samples obtained with the 16S rRNA gene primers were approximately two orders of magnitude higher than the copy numbers obtained with the mcrA primers. The newly designed mcrA primers are highly specific, whereas the 16S rRNA gene primers used in this study have the potential to amplify sequences from the whole GOM Arc I clade, possibly capturing a larger diversity of sequences that are less related to ‘Candidatus M. nitroreducens’. The target specificity was reflected in the sequence diversity: the sequenced PCR products obtained with the qPCR primer combination McrA159F/McrA345R all corresponded to the ‘Candidatus M. nitroreducens’ mcrA gene (97–100% identity at the amino acid level), whereas the sequenced PCR products of the 16S rRNA gene also included sequences (9%) that could be identified as closely related methanogens. This difference in specificity further suggests that the results obtained with these 16S rRNA gene PCR primers may overestimate the copy numbers of ‘Candidatus M. nitroreducens’ in the environment. Overall, the mcrA primers were more specific, and qPCR quantification of mcrA copy numbers may more accurately reflect the number of ‘Candidatus M. nitroreducens’ cells in a specific environment.
Among the different environments, ‘Candidatus M. nitroreducens’ was most abundant in rice field soil, followed by wastewater treatment plant sludge. The lowest copy numbers were obtained in the investigated river sediments (Fig. 2). In a previous study (Ding et al. 2015), 16S rRNA gene primers were designed to quantify ‘Candidatus Methanoperedens nitroreducens’ in two lake sediments, a river sediment, and a rice field soil sample. In that study, the total abundance of 16S rRNA gene copy numbers in rice field soil was one to two orders of magnitude lower than that obtained in the present study (3.72 × 104 to 2.30 × 105 copies μg−1 DNA versus 1.7 ± 0.4 × 106 copies μg−1 DNA in this study). This variation may be due to differences in the environmental samples used; in addition, the 16S rRNA gene primers used in that study may have been more species-specific. Importantly, the relatively high gene copy numbers obtained in both studies suggest that these anaerobic methanotrophic archaea play a significant role in mediating nitrate-dependent AOM in rice fields and contribute to mitigating methane emissions to the atmosphere.
For accurate phylogenetic analysis, only a few ‘Candidatus M. nitroreducens’ mcrA gene sequences with lengths greater than 500 bp are available in public databases. These sequences were derived from deep groundwater (Nyyssonen et al. 2012), paddy fields (Bao et al. 2014), river sediments (Jiang et al. 2011), and lake sediments (GenBank accession number JQ080004, unpublished). All of these sequences were retrieved with the general mcrA primer pair ME1F/ME2R, which yields a sequence length of 763 bp (Hales et al. 1996). These primers have a high number of mismatches with the two available full-length ‘Candidatus M. nitroreducens’ mcrA sequences: six mismatches in the forward primer and five in the reverse primer. Thus, the presence of these microorganisms and their diversity in environmental studies may be underestimated because presently used primers simply do not capture them. These archaea have been assumed to be freshwater microorganisms, and thus, it is even more remarkable that we amplified both 16S rRNA and mcrA gene sequences of ‘Candidatus M. nitroreducens’ from marine North Sea sediment. The NCBI database contains only a few sequence entries from marine samples, e.g., accession number HM746653 (unpublished) and accession number GU182109 (Lever et al. 2013), which were detected in the sediment of the Gulf of Mexico and Juan de Fuca Ridge Flank basalt seafloor sediment, respectively. The sequences have 92 and 90% identity at the nucleotide level to the mcrA gene of ‘Candidatus M. nitroreducens’ (LKCM01000102.1), respectively. For comparison, the nitrite-dependent AOM bacterium ‘Candidatus M. oxyfera’ was reported in a recent study of the Eastern South Pacific oxygen minimum zone off Chile (Padilla et al. 2016). ‘Candidatus M. oxyfera’ had previously been solely linked to freshwater environments. However, it seems that both nitrite-dependent bacteria and nitrate-dependent archaea also have niches in marine ecosystems, and their roles in these environments remain to be elucidated.
In contrast to universal mcrA primers, universal 16S rRNA gene primers have successfully captured ‘Candidatus M. nitroreducens’ sequences with high identity to ‘Candidatus M. nitroreducens’ in several environments such as minerotrophic fens (Cadillo-Quiroz et al. 2008), river sediments (Li et al. 2012; Rastogi et al. 2009), lake sediments (Kadnikov et al. 2012; Schubert et al. 2011; Stein et al. 2001), contaminated soils (Kasai et al. 2005), groundwater (Flynn et al. 2013), mud volcanoes (Wrede et al. 2012), and Antarctic cold seeps (Niemann et al. 2009), among other environments. Based on 109 sequences of the GOM Arc I group in ARB, the phylogenetic trees not only show that the sequences of this phylogenetic group form a distinct cluster but also indicate that their diversity can be further divided into sub-branches within the cluster (Welte et al. 2016). This diversity is partially correlated with the environments from which the sequences were retrieved. Due to the lack of suitable primers, there are insufficiently high-quality mcrA sequences available to perform a similar analysis. This study added 28 long ‘Candidatus M. nitroreducens’ sequences (1191 bp) suitable for high-resolution phylogenetic analysis (Fig. 3). Additional sequences are needed to confirm the splitting of the mcrA gene diversity of ‘Candidatus M. nitroreducens’ into sub-branches. Furthermore, additional mcrA gene sequences will permit an investigation of the possible link between the phylogeny and distribution of ‘Candidatus M. nitroreducens’ in nature.
In this study, we designed two novel primer sets targeting the mcrA gene of the anaerobic methanotroph ‘Candidatus M. nitroreducens’: one set suitable for quantification and the other for detailed phylogeny. These molecular tools will enable the quantification and classification of these recently discovered anaerobic microorganisms in nature and, in turn, facilitate the further elucidation of the role of this important group of archaea in global nitrogen and methane cycling.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410. doi:10.1016/S0022-2836(05)80360-2
Arshad A, Speth DR, de Graaf RM, Op den Camp HJ, Jetten MS, Welte CU (2015) A metagenomics-based metabolic model of nitrate-dependent anaerobic oxidation of methane by methanoperedens-like archaea. Front Microbiol 6:1423. doi:10.3389/fmicb.2015.01423
Bao QL, Xiao KQ, Chen Z, Yao HY, Zhu YG (2014) Methane production and methanogenic archaeal communities in two types of paddy soil amended with different amounts of rice straw. FEMS Microbiol Ecol 88(2):372–385. doi:10.1111/1574-6941.12305
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2013) GenBank. Nucleic Acids Res 41(Database issue):D36–D42. doi:10.1093/nar/gks1195
Cadillo-Quiroz H, Yashiro E, Yavitt JB, Zinder SH (2008) Characterization of the archaeal community in a minerotrophic fen and terminal restriction fragment length polymorphism-directed isolation of a novel hydrogenotrophic methanogen. Appl Environ Microbiol 74(7):2059–2068. doi:10.1128/aem.02222-07
Deutzmann JS, Schink B (2011) Anaerobic oxidation of methane in sediments of an oligotrophic freshwater lake (Lake Constance). Appl Environ Microbiol. doi:10.1128/aem.00340-11
Ding J, Ding ZW, Fu L, Lu YZ, Cheng SH, Zeng RJ (2015) New primers for detecting and quantifying denitrifying anaerobic methane oxidation archaea in different ecological niches. Appl Microbiol Biotechnol. doi:10.1007/s00253-015-6893-6
Egger M, Rasigraf O, Sapart CJ, Jilbert T, Jetten MS, Rockmann T, van der Veen C, Banda N, Kartal B, Ettwig KF, Slomp CP (2015) Iron-mediated anaerobic oxidation of methane in brackish coastal sediments. Environ Sci Technol 49(1):277–283. doi:10.1021/es503663z
Ettwig KF, Shima S, van de Pas-Schoonen KT, Kahnt J, Medema MH, Op den Camp HJ, Jetten MS, Strous M (2008) Denitrifying bacteria anaerobically oxidize methane in the absence of archaea. Environ Microbiol 10(11):3164–3173. doi:10.1111/j.1462-2920.2008.01724.x
Ettwig KF, van Alen T, van de Pas-Schoonen KT, Jetten MS, Strous M (2009) Enrichment and molecular detection of denitrifying methanotrophic bacteria of the NC10 phylum. Appl Environ Microbiol 75(11):3656–3662. doi:10.1128/AEM.00067-09
Ettwig KF, Butler MK, Le Paslier D, Pelletier E, Mangenot S, Kuypers MMM, Schreiber F, Dutilh BE, Zedelius J, de Beer D, Gloerich J, Wessels HJCT, van Alen T, Luesken F, Wu ML, van de Pas-Schoonen KT, Op den Camp HJM, Janssen-Megens EM, Francoijs KJ, Stunnenberg H, Weissenbach J, Jetten MSM, Strous M (2010) Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464(7288):543–548. doi:10.1038/nature08883
Flynn TM, Sanford RA, Ryu H, Bethke CM, Levine AD, Ashbolt NJ, Santo Domingo JW (2013) Functional microbial diversity explains groundwater chemistry in a pristine aquifer. BMC Microbiol 13:146. doi:10.1186/1471-2180-13-146
Hales BA, Edwards C, Ritchie DA, Hall G, Pickup RW, Saunders JR (1996) Isolation and identification of methanogen-specific DNA from blanket bog feat by PCR amplification and sequence analysis. Appl Environ Microbiol 62(2):668–675
Hallam SJ, Girguis PR, Preston CM, Richardson PM, Delong EF (2003) Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane-oxidizing archaea. Appl Environ Microbiol 69(9):5483–5491
Hallam SJ, Putnam N, Preston CM, Detter JC, Rokhsar D, Richardson PM, DeLong EF (2004) Reverse methanogenesis: testing the hypothesis with environmental genomics. Science 305:1457–1462
Haroon MF, Hu S, Shi Y, Imelfort M, Keller J, Hugenholtz P, Yuan Z, Tyson GW (2013) Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500(7464):567–570
Hinrichs KU, Boetius A (2002) The anaerobic oxidation of methane: new insights in microbial ecology and biogeochemistry. In: Wefer G, Billett D, Hebbeln D, Jørgensen BB, Schlüter M, van Weering T (eds) Ocean margin systems. Springer, Heidelberg, pp. 457–477
Hu BL, Shen LD, Lian X, Zhu Q, Liu S, Huang Q, He ZF, Geng S, Cheng DQ, Lou LP, Xu XY, Zheng P, He YF (2014) Evidence for nitrite-dependent anaerobic methane oxidation as a previously overlooked microbial methane sink in wetlands. Proc Natl Acad Sci U S A 111(12):4495–4500. doi:10.1073/pnas.1318393111
Jiang L, Zheng Y, Chen J, Xiao X, Wang F (2011) Stratification of archaeal communities in shallow sediments of the Pearl River Estuary, Southern China. Antonie Van Leeuwenhoek 99(4):739–751. doi:10.1007/s10482-011-9548-3
Juottonen H, Galand PE, Yrjala K (2006) Detection of methanogenic archaea in peat: comparison of PCR primers targeting the mcrA gene. Res Microbiol 157(10):914–921. doi:10.1016/j.resmic.2006.08.006
Kadnikov VV, Mardanov AV, Beletsky AV, Shubenkova OV, Pogodaeva TV, Zemskaya TI, Ravin NV, Skryabin KG (2012) Microbial community structure in methane hydrate-bearing sediments of freshwater Lake Baikal. FEMS Microbiol Ecol 79(2):348–358. doi:10.1111/j.1574-6941.2011.01221.x
Kasai Y, Takahata Y, Hoaki T, Watanabe K (2005) Physiological and molecular characterization of a microbial community established in unsaturated, petroleum-contaminated soil. Environ Microbiol 7(6):806–818
Knittel K, Boetius A (2009) Anaerobic oxidation of methane: progress with an unknown process. Annu Rev Microbiol 63:311–334
Knittel K, Lösekann T, Boetius A, Kort R, Amann R (2005) Diversity and distribution of methanotrophic archaea at cold seeps. Appl Environ Microbiol 71(1):467–479
Krüger M, Meyerdierks A, Glöckner FO, Amann R, Widdel F, Kube M, Reinhardt R, Kahnt J, Böcher R, Thauer RK, Shima S (2003) A conspicuous nickel protein in microbial mats that oxidize methane anaerobically. Nature 426(18):878–881
Lever MA, Rouxel O, Alt JC, Shimizu N, Ono S, Coggon RM, Shanks WC 3rd, Lapham L, Elvert M, Prieto-Mollar X, Hinrichs KU, Inagaki F, Teske A (2013) Evidence for microbial carbon and sulfur cycling in deeply buried ridge flank basalt. Science 339(6125):1305–1308. doi:10.1126/science.1229240
Li Q, Wang F, Chen Z, Yin X, Xiao X (2012) Stratified active archaeal communities in the sediments of Jiulong River estuary, China. Front Microbiol 3:311. doi:10.3389/fmicb.2012.00311
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar BA, Lai T, Steppi S, Jobb G (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371. doi:10.1093/nar/gkh293
Luesken FA, Zhu BL, van Alen TA, Butler MK, Diaz MR, Song B, den Camp HJMO, Jetten MSM, Ettwig KF (2011) pmoA primers for detection of anaerobic methanotrophs. Appl Environ Microbiol 77(11):3877–3880. doi:10.1128/Aem.02960-10
Luton PE, Wayne JM, Sharp RJ, Riley PW (2002) The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148(11):3521–3530
Mills HJ, Hodges C, Wilson K, MacDonald IR, Sobecky PA (2003) Microbial diversity in sediments associated with surface-breaching gas hydrate mounds in the Gulf of Mexico. FEMS Microbiol Ecol 46(1):39–52. doi:10.1016/s0168-6496(03)00191-0
Myhre G, Shindell D, Bréon FM, Collins W, Fuglestvedt J, Huang J, Koch D, Lamarque JF, Lee D, Mendoza B, Nakajima T, Robock A, Stephens G, Takemura T., Zhang H (2013) Anthropogenic and natural radiative forcing. In: Climate change 2013: the physical science basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change Cambridge University Press: Cambridge
Nauhaus K, Treude T, Boetius A, Krüger M (2005) Environmental regulation of the anaerobic oxidation of methane: a comparison of ANME-I and ANME-II communities. Environ Microbiol 7(1):98–106
Niemann H, Fischer D, Graffe D, Knittel K, Montiel A, Heilmayer O, Nothen K, Pape T, Kasten S, Bohrmann G, Boetius A, Gutt J (2009) Biogeochemistry of a low-activity cold seep in the Larsen B area, western Weddell Sea, Antarctica. Biogeosciences 6(11):2383–2395
Nunoura T, Oida H, Toki T, Ashi J, Takai K, Horikoshi K (2006) Quantification of mcrA by quantitative fluorescent PCR in sediments from methane seep of the Nankai Trough. FEMS Microbiol. Ecol. 57:149–157. doi:10.1111/j.1574-6941.2006.00101.x
Nunoura T, Oida H, Miyazaki J, Miyashita A, Imachi H, Takai K (2008) Quantification of mcrA by fluorescent PCR in methanogenic and methanotrophic microbial communities. FEMS Microbiol Ecol 64(2):240–247. doi:10.1111/j.1574-6941.2008.00451.x
Nyyssonen M, Bomberg M, Kapanen A, Nousiainen A, Pitkanen P (2012) Methanogenic and sulphate reducing communities in deep groundwater from crystalline rock fractures in Olkiluoto, Finland. Geomicrobiol J 29(10):863–878
Padilla CC, Bristow LA, Sarode N, Garcia-Robledo E, Gomez Ramirez E, Benson CR, Bourbonnais A, Altabet MA, Girguis PR, Thamdrup B, Stewart FJ (2016) NC10 bacteria in marine oxygen minimum zones. ISME J. doi:10.1038/ismej.2015.262
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(Database issue):D590–D596. doi:10.1093/nar/gks1219
Raghoebarsing AA, Pol A, van de Pas-Schoonen KT, Smolders AJ, Ettwig KF, Rijpstra WI, Schouten S, Damste JS, Op den Camp HJ, Jetten MS, Strous M (2006) A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 440(7086):918–921. doi:10.1038/nature04617
Rastogi G, Sani RK, Peyton BM, Moberly JG, Ginn TR (2009) Molecular studies on the microbial diversity associated with mining-impacted Coeur d’Alene river sediments. Microb Ecol 58(1):129–139
Schubert CJ, Vazquez F, Lösekann-Behrens T, Knittel K, Tonolla M, Boetius A (2011) Evidence for anaerobic oxidation of methane in sediments of a freshwater system (Lago di Cadagno). FEMS Microbiol Ecol 76(1):26–38. doi:10.1111/j.1574-6941.2010.01036.x
Schwietzke S, Sherwood OA, Bruhwiler LMP, Miller JB, Etiope G, Dlugokencky EJ, Michel SE, Arling VA, Vaughn BH, White JWC, Tans PP (2016) Upward revision of global fossil fuel methane emissions based on isotope database. Nature 538(7623):88–91. doi:10.1038/nature19797
Springer E, Sachs MS, Woese CR, Boone DR (1995) Partial Gene Sequences for the A Subunit of Methyl-Coenzyme M Reductase (mcrI) as a Phylogenetic Tool for the Family Methanosarcinaceae. Int J of Syst Bacteriol 45(3):554–559
Stadnitskaia A, Muyzer G, Abbas B, Coolen MJL, Hopmans EC, Baas M, van Weering TCE, Ivanov MK, Poludetkina E, Damste JSS (2005) Biomarker and 16S rDNA evidence for anaerobic oxidation of methane and related carbonate precipitation in deep-sea mud volcanoes of the Sorokin Trough, Black Sea. Mar Geol 217(1–2):67–96. doi:10.1016/j.margeo.2005.02.023
Stein LY, La Duc MT, Grundl TJ, Nealson KH (2001) Bacterial and archaeal populations associated with freshwater ferromanganous micronodules and sediments. Environ Microbiol 3(1):10–18
Vaksmaa A, Luke C, van Alen T, Vale G, Lupotto E, Jetten MS, Ettwig KF (2016) Distribution and activity of the anaerobic methanotrophic community in a nitrogen-fertilized Italian paddy soil. FEMS Microbiol Ecol 92(12). doi:10.1093/femsec/fiw181
Welte C, Rasigraf O, Vaksmaa A, Versantvoort W, Arshad A, Op den Camp H, Jetten M, Luke C, Reimann J (2016) Nitrate-and nitrite-dependent anaerobic oxidation of methane. Environmental Microbiology and Environmental Microbiology Reports
Wrede C, Brady S, Rockstroh S, Dreier A, Kokoschka S, Heinzelmann SM, Heller C, Reitner J, Taviani M, Daniel R, Hoppert M (2012) Aerobic and anaerobic methane oxidation in terrestrial mud volcanoes in the Northern Apennines. Sediment Geol 263-264:210–219. doi:10.1016/j.sedgeo.2011.06.004
Zhou L, Wang Y, Long XE, Guo J, Zhu G (2014) High abundance and diversity of nitrite-dependent anaerobic methane-oxidizing bacteria in a paddy field profile. FEMS Microbiol Lett 360(1):33–41. doi:10.1111/1574-6968.12567
Zhu B, van Dijk G, Fritz C, Smolders AJ, Pol A, Jetten MS, Ettwig KF (2012) Anaerobic oxidization of methane in a minerotrophic peatland: enrichment of nitrite-dependent methane-oxidizing bacteria. Appl Environ Microbiol 78(24):8657–8665. doi:10.1128/AEM.02102-12
Acknowledgments
We thank Rienke F. Uijen (Radboud University, Nijmegen, NL) for carrying out initial primer testing during her internship and Ramesh Goel (Utah University, Salt Lake City, USA) for providing the environmental samples of Jordan River sediment and State Channel sediment.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This work was supported by the Netherlands Organization for Scientific Research [VENI 863.13.007 to KFE], the European Research Council [ERC AG 339880 Eco_MoM to MSMJ, AV and CL], the Gravitation grant [024002002 Soehngen Institute of Anaerobic Microbiology to MSMJ; 024002001 NESSC], and the Spinoza prize to MSMJ.
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights and informed consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 314 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vaksmaa, A., Jetten, M.S.M., Ettwig, K.F. et al. McrA primers for the detection and quantification of the anaerobic archaeal methanotroph ‘Candidatus Methanoperedens nitroreducens’. Appl Microbiol Biotechnol 101, 1631–1641 (2017). https://doi.org/10.1007/s00253-016-8065-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-8065-8