
Abstract
Despite substantial progress, mortality and morbidity of the acute respiratory distress syndrome (ARDS), a severe form of acute lung injury (ALI), remain unacceptably high. There is no effective treatment for ARDS/ALI. The renin–angiotensin system (RAS) through Angiotensin-converting enzyme (ACE)-generated Angiotensin II contributes to lung injury. ACE2, a recently discovered ACE homologue, acts as a negative regulator of the RAS and counterbalances the function of ACE. We hypothesized that ACE2 prevents Bleomycin (BLM)-induced lung injury. Fourteen to 16-week-old ACE2 knockout mice—male (ACE2−/y) and female (ACE2−/−)—and age-matched wild-type (WT) male mice received intratracheal BLM (1.5U/kg). Male ACE2−/y BLM injured mice exhibited poorer exercise capacity, worse lung function and exacerbated lung fibrosis and collagen deposition compared with WT. These changes were associated with increased expression of the profibrotic genes α-smooth muscle actin (α-SMA) and Transforming Growth Factor ß1. Compared with ACE2−/y exposed to BLM, ACE2−/− exhibited better lung function and architecture and decreased collagen deposition. Treatment with intraperitoneal recombinant human (rh) ACE2 (2 mg/kg) for 21 days improved survival, exercise capacity, and lung function and decreased lung inflammation and fibrosis in male BLM-WT mice. Female BLM WT mice had mild fibrosis and displayed a possible compensatory upregulation of the AT2 receptor. We conclude that ACE2 gene deletion worsens BLM-induced lung injury and more so in males than females. Conversely, ACE2 protects against BLM-induced fibrosis. rhACE2 may have therapeutic potential to attenuate respiratory morbidity in ALI/ARDS.
Similar content being viewed by others

Introduction
Acute respiratory distress syndrome (ARDS), a severe form of acute lung injury (ALI), is characterized by diffuse lung damage, arterial hypoxaemia, and decreased compliance [1]. Despite advances in patient care, ALI/ARDS remains an important cause of death and morbidity in intensive care units [2]. ARDS survivors often have impaired pulmonary function, decreased health-related quality of life, and chronic pulmonary fibrosis [3–5]. At present, treatment is supportive and includes prolonged mechanical ventilation, which can further contribute to ARDS-related morbidity [6]. Currently, there is no effective pharmacological therapy to attenuate lung injury and promote lung repair in ALI/ARDS [6–10].
Experimental and clinical evidence indicate that the renin–angiotensin system (RAS) contributes to lung injury [11–14]. ACE cleaves angiotensin (ANG) I to generate ANGII. Proliferative and fibrotic properties have been attributed to the ACE-ANGII-AT1 receptor axis [15, 16]. ACE2, a homologue to ACE, degrades ANGII to ANG 1–7 peptides, limiting ANGII accumulation [12, 14]. Thus, ACE2 counterbalances the deleterious effects of ACE and prevents lung injury. Angiotensin converting enzyme-2 (ACE2) knockout mice have enhanced vascular permeability, increased lung edema, neutrophil accumulation, and worsened lung function [12]. Characteristics similar to those seen in lungs of patients with ARDS have been modeled in mice exposed to bleomycin (BLM)-induced lung injury complicated by fibrosis [17]. We hypothesized that ACE2 gene deletion would worsen BLM-induced lung injury and its resultant fibrosis; conversely, recombinant human (rh) ACE2 would attenuate BLM-induced lung injury. Furthermore, since lungs of female rats exhibit higher lung expression of ACE2 than male rats [18], we assessed gender differences in ACE2 knockout mice in BLM-induced lung injury.
Materials and methods
Experimental design
All procedures involving animals were approved by the Animal Welfare Committee of the University of Alberta. Fourteen to 16-week-old male C57BL6 (wild type, WT, controls) mice and ACE2 knockouts—males (ACE2−/y) and females (ACE2−/−)—were allocated to the following groups: (1) WT control group (saline), (2) ACE2−/y control group (saline), (3) WT BLM (ALI/ARDS model), and (4) ACE2−/y BLM. The mice were anesthetized using a mixture of oxygen and Isofluorane® 2.5%, weighted and BLM (Sigma Aldrich, St. Louis, MO) administered intratracheally at a dose of 1.5 U/kg in 50 μl (∼0.04 U/mice) [19]. Control animals were injected intratracheally with 50 μl of sterile saline 0.9%.
In a separate set of experiments, we compared gender differences in the following groups: (1) ACE2−/y controls (saline), (2) ACE2−/− controls, (3) ACE2−/y BLM, and (4) ACE2−/− BLM.
Finally, to explore the therapeutic potential of rhACE2, we performed a third set of experiments including (1) WT control group (saline), (2) WT BLM (ALI/ARDS model), and (3) WT BLM+rhACE2. rhACE2 was given intraperitoneally for 21 days at a dose of 2 mg/kg.
Mice were monitored every day. An exercise test was performed 21 days after BLM or saline injection. Three weeks after BLM, lungs were harvested for hydroxyproline and collagen assays, profibrotic gene expression, and histology, after assessing lung mechanics.
Recombinant human ACE2
The extracellular domain of rhACE2 (amino acid residues 1–740, MW = 101 kDa) was expressed recombinantly in Chinese hamster ovary cells under serum free conditions in a chemically defined medium as previously described [20–23]. The enzymatic turnover of rhACE2 with Ang II substrate was 5.2 ± 0.1 μmol mg−1 min−1 and the elimination half-life of rhACE2 was 10.4 h in Rhesus monkeys. The purity of the expression product was 99.99% as measured by HPLC [21, 22].
TaqMan real-time polymerase chain reaction
mRNA expression levels of smooth muscle actin and transforming growth factor-β were determined by TaqMan (Applied Systems Inc, Streetsville, Ontario, Canada) real-time polymerase chain reaction as described previously [24–26]. Expression analysis of the reported genes was performed by TaqMan reverse-transcription polymerase chain reaction using ABI 7900 sequence detection system; 18S rRNA was used as an endogenous control. The primer/probe for mRNA expression analysis by Taqman real-time polymerase chain reaction were purchased from Applied Biosystems for α-smooth muscle actin (α-SMA; product #: Mm00725412_S1) and as follows for (Transforming Growth Factor ß1) TGFß1 Forward Primer 5′-CCTGCAAGACCATCGACATG-3′; Reverse Primer 5′-ACAGGATCTGGCCACGGAT-3′ and Probe 5′-FAMCTGGTGAAACGGAAGCGCATCGAA-TAMRA -3′. Results are presented as relative expression to 18S.
Exercise capacity
The animals run on a treadmill with an inclination of 5° at different speeds/time as follows: 1 min at 3, 4, and 5 m/min respectively and 3 min at 6 m/min, as a warm up. This followed by 24 min at 8 m/min, 7 min at 10 m/min, and 8 min at 12 m/min. Mice were run until they could not maintain sufficient speed to remain off the shock grid [27].
Lung function testing
Mice were anesthetized 21 days after BLM administration with 70–90 mg/kg pentobarbital sodium, tracheotomized, and mechanically ventilated at a rate of 350 breaths per minute, tidal volume of 6 ml/kg, and positive end-expiratory pressure of 3–4 cm H2O with a computer-controlled small-animal ventilator (Scireq, Montreal, Canada). Once ventilated, mice were paralyzed with 1 mg/kg pancuronium bromide i.p. (Sigma Aldrich, St. Louis, MO) to evaluate lung mechanics and record airway pressure, volume, and airflow using a controlled piston.
Pressure–volume curves were generated by a sequential and increasing delivery of air into the lungs from resting pressure (zero volume) to total lung capacity followed by sequential expiratory steps during which air was incrementally released. The plateau pressure was recorded when airflow returned to zero at each step. To determine compliance of the lung, the Salazar–Knowles equation was applied to the pressure measurements obtained between total lung capacity and functional residual capacity during the expiratory phase of the pressure–volume loop [28]. Compliance was determined form the analysis of pressure-volume curves.
Forced oscillation technique measures the impedance (alveolar pressure to central airflow ratios) of the lung to an oscillatory flow wave controlled by the computer piston. These impedance values are applied to a mathematical model of the lung, the constant phase model [29]. This model provides a clear distinction between central and peripheral airways and lung parenchyma. The equation of the constant phase model is
where R aw = a measure of central airways resistance, I aw = inertance of the gas in the airways, G ti = tissue resistance, H ti = tissue elasticity, and i = √−1. The computer-controlled piston applies a 1.25 to 8-s perturbation to the lungs consisting of priming frequencies from 1 to 20.5 Hz. Multiple linear regression is used to fit impedance spectra derived from measured pressure and volume changes to the constant phase model of the lung.
To ensure proper recruitment of all alveolar spaces, three pressure–volume curves were generated for each animal. After this maneuver, a 3-s prime wave was performed followed by a second pressure–volume curve to obtain reported values. Each perturbation was followed by 10 s of ventilation before the next measurement was taken [28, 29].
Lung histology
The right bronchus was ligated and the left lung inflated with 10% formaldehyde at a pressure of 20 cm H2O for histology. Inflation fixed lungs were then embedded in paraffin, sectioned as described [27] and stained with Trichrome Masson’s; muscles and cells are stained red, nuclei black, and collagen blue. For histologic evaluation of the lungs, four midlung sections per lung were examined.
Hydroxyproline assay
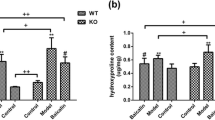
Mice lungs were harvested 21 days after BLM/saline administration and perfused with heparinized saline to remove blood. The right bronchus was ligated and each of the four right lobes was weighted, snap frozen in liquid nitrogen, and stored at −80°C, for hydroxyproline and total collagen content assays. Briefly, lungs were homogenized, incubated in 50% trichloroacetic acid (Sigma Aldrich, St. Louis, MO) and hydrolyzed with 12N HCl. Samples were then baked at 110°C for 12 h. Hydroxyproline is oxidized using chloramine T (Fluka,) and pink colored with Ehrlich’s solution (Sigma, St. Louis, MO). The concentration of hydroxyproline is calculated against a hydroxyproline standard curve and the values of the samples are normalized to dry tissue weight, expressed as μg/mg [30, 31].
Lung collagen content
Collagen content of the lung was determined by assaying soluble collagen using the Sircol Collagen Assay kit, a modification of the sirius red method (Biocolor, Belfast, Northern Ireland), according to the manufacturer’s instructions. Briefly, frozen samples were thawed, incubated at 4°C overnight in lysis buffer according to tissue weight (0.5 M acetic acid and protein inhibitor cocktail, Sigma Aldrich, St. Louis, MO). Supernatants (50 μl) were added to 1 ml of Sircol Dye Reagent and then mixed for 30 min at room temperature in a mechanical shaker. The collagen–dye complex was precipitated by centrifugation at 10,000×g for 10 min. The unbound dye solution was carefully removed. The precipitated complex was resuspended in 1 ml of alkali reagent. The obtained solution was placed in a 96-wells plate and evaluated in a plate reader (absorbance = 540 nm). Obtained values were then compared to the standard curve as recommended to obtain absolute collagen content. Shown data represent the mean collagen content expressed as μg/mg.
Western blot
Right lungs were flash-frozen in liquid nitrogen and homogenized in buffer containing an antiprotease cocktail before electrophoresis on 7.5% or 10% SDS-PAGE gels. Protein was quantified using the Bradford method. Lung expression of AT1 and AT2 receptors were quantified by densitometry, relative to a reporter (actin, 43 kDa). AT1 and AT2 primary antibodies were purchased from Santa Cruz.
ELISA
Right lungs were flash-frozen in liquid nitrogen and homogenized in buffer containing an antiprotease cocktail before quantitative assessment of proinflamatory cytokines keratinocyte chemoattractant, tumor necrosis factor alpha (TNFα), interleukin 1β, and TGFβ were performed using ELISA kits (R&D Systems).
Statistical analysis
Data are expressed as mean ± SE, except where stated otherwise. Statistical analysis was performed using unpaired Student’s t test or ANOVA, post hoc test (least significant difference), and Mann–Whitney as appropriate. Values were considered significant with P < 0.05.
Results
Loss of ACE2 worsens lung fibrosis in BLM-induced lung injury
The severity of lung fibrosis between groups was assessed qualitatively on Trichrome Masson’s stained lung sections (Fig. 1a) and quantitatively by measuring mRNA levels of profibrotic genes α-SMA (Fig. 1b) and TGFβ1 (Fig. 1c) as well as total lung collagen/hydroxyproline content (Fig. 1d, e). As expected, no histological changes were observed in the lungs from saline (controls) treated WT and ACE2−/y animals (Fig. 1a). BLM-induced loss of lung architecture and fibrosis in the parenchyma (Fig. 1a) were more evident in ACE2−/y-BLM compared with controls and BLM-WT mice. These changes were associated with increased expression of the fibrosis-associated genes α-SMA and TGFβ1 (Fig. 1b-c) in both BLM treated groups. Collagen and hydroxyproline levels were significantly higher in ACE2−/y BLM compared with controls and BLM-WT (Fig. 1d, e).

ACE2 deletion worsens lung fibrosis in BLM-induced lung injury. a Representative Trichrome Masson’s histological sections. BLM-induced lung injury, evidenced by loss of lung architecture and collagen deposition (blue), is more evident in BLM-ACE2−/y compared with BLM-WT mice. b, c Lung α SMA and TGFβ1 mRNA are significantly increased in BLM ACE2−/y and WT BLM compared with WT and ACE2−/y controls. RE relative exposure to 18S. d, e Lung collagen and hydroxyproline content were significantly increased in BLM ACE2−/y compared with WT and controls. Mean data ± SEM. N = 4–7/group, *p < 0.05
Loss of ACE2 worsens exercise capacity and lung function in BLM-induced lung injury
After 21 days of BLM injection, all BLM exposed animals (ACE2−/y and WT) exhibited a significant decrease in exercise capacity compared with saline-treated control mice (Fig. 2). Exercise capacity was significantly worse in BLM-ACE2−/y compared with BLM-WT. Likewise, BLM-ACE2−/y mice exhibited decreased dynamic lung compliance (Fig. 3a) and increased lung elastance (Fig. 3b) compared with BLM-WT.

Exercise capacity in BLM-induced lung injury in WT and ACE2−/y. ACE2 deletion worsens exercise capacity 21 days after BLM exposure. Percentage change respective to controls (male WT and ACE2−/y unexposed to BLM) ± SEM. N = 5–7/group, *p < 0.05

Changes in lung mechanics at 21 days in BLM-induced fibrosis in WT and ACE2−/y. a Dynamic compliance is decreased in BLM challenged ACE2−/y mice compared with BLM-WT. Data are expressed as percentage change respective to controls (male WT and ACE2−/y unexposed to BLM) ± SEM. b Dynamic Elastance is increased in ACE2−/y mice compared with BLM-WT, data expressed as percentage change respective to controls ± SEM. N = 4–7/group, *p < 0.05
Male ACE2−/y display worse lung fibrosis than female ACE2−/− in BLM-induced lung injury
Trichrome Masson’s stained lung sections showed loss of lung architecture and increased fibrosis in BLM-ACE2−/y as compared with saline controls and BLM-ACE2−/− (Fig. 4a). Lung fibrosis appeared significantly milder in BLM-ACE2−/− females. BLM-ACE2−/y had significantly higher levels of α-SMA (Fig. 4b) and TGFβ1 (Fig. 4c) mRNA and collagen deposition compared with ACE2−/− BLM (Fig. 4d, e). Likewise, these results were associated with decreased compliance and increased elastance in BLM-ACE2−/y compared with female BLM-ACE2−/− mice (Fig. 5a, b).

Gender differences in response to BLM-induced lung injury. a Representative Trichrome Masson’s histological sections. BLM ACE2−/− shows milder disruption of lung architecture as compared with BLM ACE2−/y group. b, c Lung α SMA and TGFβ1 mRNA are significantly decreased in BLM ACE2−/− compared with BLM ACE2−/y. Mean data ± SEM. RE relative exposure to 18S. d, e Lung total collagen and hydroxyproline content are increased in BLM ACE2−/y compared with BLM ACE2−/−. Mean data ± SEM. N = 4–7/group, *p < 0.05

Lung function in ACE2 knockout mice. a Dynamic Compliance is significantly decreased in BLM ACE2−/y compared with ACE2−/−. b Dynamic Elastance is increased in ACE2−/y BLM compared with BLM ACE2−/−. Data expressed as percentage change respective to controls (ACE2−/y and ACE2−/− unexposed to BLM) ± SEM. N = 5–7/group, *p < 0.05
Treatment with rhACE2 improves lung architecture and function and attenuates lung collagen deposition in BLM-induced lung injury in WT mice
We examined the protective effect of rhACE2 to prevent fibrosis in BLM-induced lung injury. Treatment with rhACE2 improved survival (Fig. 6a), exercise capacity (Fig. 6b) and lung function (dynamic compliance and elastance, Fig. 6c, d) as compared with untreated BLM-WT mice. Treatment with rhACE2 also improved lung architecture in Trichrome Masson’s stained lung sections (Fig. 7a). rhACE2 significantly decreased lung collagen deposition in BLM-WT+rhACE2 compared with BLM-WT (Fig. 7b), as well as levels of α-SMA (Fig. 7c) and TGFβ1 mRNA (Fig. 7d) and protein (Fig. 7e) and lung TNFα protein expression (Fig. 7f) as compared with untreated BLM-WT mice.

a Cumulative survival of BLM-WT mice and BLM WT+rhACE2 treated mice. rhACE2 treatment significantly improved survival compared with untreated BLM-WT mice. N = 12–15/group, *p < 0.05. b rhACE2 treatment improves exercise capacity compared with untreated WT BLM mice. c, d rhACE2 treatment improves lung function: dynamic compliance is significantly increased in BLM-WT+rhACE2 compared with BLM-WT; Dynamic Elastance is significantly decreased in BLM-WT+rhACE2. Data expressed as percentage change respective to controls (male WT unexposed to BLM) ± SEM. N = 4–7/group, *p < 0.05

a Representative Trichrome Masson’s histological sections. BLM-WT+rhACE2 showed improved lung architecture as compared with BLM-WT mice. b Lung total collagen content and hydroxyproline are decreased in BLM-WT+rhACE2 compared with untreated BLM-WT mice. Mean data ± SEM. c–f Lung α SMA and TGFβ1 mRNA and protein as well as TNFα are significantly decreased in BLM-WT+rhACE2 compared with WT BLM mice. Mean data ± SEM. RE relative exposure to 18S
Effects of rhACE2 in male and female BLM-WT mice on lung architecture, function and lung collagen deposition WT-treated mice
Finally, we examined the protective effect of rhACE2 to prevent fibrosis in female BLM-induced lung injury (Fig. 8). Surprisingly, BLM challenged WT females did not exhibit fibrotic changes. Trichrome Masson’s stained lung sections revealed mild lung architecture distorsion in BLM WT females compared with males (Fig. 8a). WT females exposed to BLM had only mild increase in lung collagen (Fig. 8b) and lung hydroxyproline (Fig. 8c) compared with males. Likewise, WT females had unchanged lung function (Fig. 8d, e) and exercise capacity (Fig. 8f) compared with BLM WT males. rhACE2 significantly attenuated lung fibrosis (Fig. 8a–c) and improved lung function (Fig. 8d, e) and exercise capacity (Fig. 8f) in BLM WT males.

a Representative Trichrome Masson’s histological sections. BLM-female WT showed decreased lung architecture distortion as compared with BLM-male WT mice. Moreover, rhACE2 further improved lung architecture in BLM-female WT. b, c Lung total collagen content and hydroxyproline are decreased in BLM-female WT, BLM-female WT+rhACE2, and BLM-male WT+rhACE2, compared with untreated BLM-male WT mice. Mean data ± SEM. d, e BLM-female WT exhibit similar lung function to untreated controls and BLM-male WT+rhACE2: dynamic compliance is significantly increased in BLM-female WT and BLM-female WT+rhACE2 compared with BLM-male WT; Dynamic Elastance is significantly decreased in BLM-female WT and BLM-female WT+rhACE2 compared with BLM-male WT. f Exercise capacity was unchanged in female treated and untreated groups compared with BLM-male WT. Data expressed as percentage change respective to controls (male and female WT unexposed to BLM) ± SEM. N = 4–7/group, *p < 0.05
Angiotensin receptors in BLM-induced lung injury
In order to explain the apparent protection of female WT mice from fibrosis, we explored other component of the RAS. Lung AT1R expression was unchanged between Control and BLM exposed WT female and WT male (Fig. 9a). Conversely, lung AT2R expression was significantly increased in BLM female WT as compared with control female WT and BLM male WT (Fig. 9b). The AT2/AT1 receptor ratio was significantly increased in BLM WT female (Fig. 9c) compared with all other groups.

a Representative immunoblots for lung AT1R and Actin (control) expression in lung homogenates from WT control males/females and BLM WT challenged male and female mice and mean densitometry for lung AT1R expression. b Representative immunoblots for lung AT2R and Actin (control) expression in lung homogenates from WT control males/females and BLM WT challenged male and female mice and mean densitometry for lung AT2R expression. c AT2/AT1 receptor ratio is significantly increased in WT BLM challenged females; ±SEM. N = 3group, *p < 0.05
Discussion
We observed that ACE2 gene deletion aggravates BLM-induced lung injury. Conversely, rhACE2 treatment improves lung function and exercise capacity and attenuates lung fibrosis in BLM-induced lung injury. In addition, we found that BLM-induced lung injury was worse in ACE2 deficient male than female mice. Overall, our data suggest a protective role for rhACE2 in experimental ALI/ARDS.
ACE2 in lung injury and repair
Recent evidence suggests that the RAS has important functions outside the cardio-vascular system. Latest since ACE2 was identified as a key receptor for coronavirus infections responsible for the severe acute respiratory syndrome [14] major attention has been drawn to the potential protective role of ACE2 in lung diseases. In three different ALI/ARDS models (acid-aspiration-induced ARDS, endotoxin-induced ARDS, and peritoneal sepsis-induced), ACE2 knockout mice exhibit exacerbated lung injury compared with WT mice [12]: loss of ACE2 caused enhanced vascular permeability, increased lung edema, neutrophil accumulation, and worsened lung function. Importantly, treatment with catalytically active recombinant ACE2 protein improved the symptoms of ALI in WT mice, as well as in ACE2 knockout mice [12]. Furthermore, lung injury in experimental ARDS in mice can be attenuated by blocking the RAS [12]. One complication of ARDS is lung fibrosis. Li et al. [32] have demonstrated that ACE2 mRNA and activity are downregulated in human and experimental lung fibrosis and suggest that ACE2 limits the local accumulation of ANG II. Previous studies have not assessed the effect of ACE2 deletion on long-term complications of ALI/ARDS such as lung fibrosis. Our data demonstrate that loss of ACE2 aggravates exercise capacity and lung function and worsens subsequent lung fibrosis in BLM-induced experimental lung injury.
Currently, there is no efficacious pharmacological therapy to prevent the onset of pulmonary fibrosis post ALI/ARDS [7]. Consequently, we explored the therapeutic potential of rhACE2 to attenuate BLM-induced lung injury. rhACE2 improved survival, exercise capacity, and lung function and abrogated lung fibrosis in this model. Our data are in accordance with recent findings showing that lentiviral packaged ANG [1–7] fusion gene or ACE2 cDNA prevents BLM-induced lung fibrosis in male Sprague Dawley rats [33]. This is also consistent with recent reports showing a protective effect of ACE2 in preventing fibrosis in other organs such as ANGII-induced cardiac hypertrophy and in diabetic nephropathy [21, 22]. Our data provide additional proof of principle for the therapeutic benefit of ACE2 in improving lung function and structure post-ALI/ARDS. The therapeutic implications of these findings could potentially translate into decreased morbidity and mortality in ALI/ARDS patients.
Gender differences in the susceptibility of ACE2 knockout mice to BLM-induced lung injury
In this study, we found gender differences in ACE2−/− mice in BLM-induced lung injury. Men with idiopathic pulmonary fibrosis have decreased quality of life compared with women [34]. In rodents, castrated male mice exhibited a female-like response to BLM while female mice given exogenous androgen exhibited a male-like response, suggesting a detrimental role of androgens in pulmonary function in fibrosis [35]. Interestingly, in BLM-induced fibrosis in rats, female hormones appeared to have a detrimental role in BLM-exposed female compared with males [36]. In our study, the effects of BLM in female ACE2−/− mice were not only milder compared with male knock-out mice, but also milder compared with male Wt mice. These contradictory results could potentially be explained by differences in species [37] and BLM doses. In our study, we selected a low to middle BLM dose in which a balance between survival and fibrotic response (functional and histological) was achieved. It is recognized that lung function correlates more directly with poor prognosis than fibrotic end-points [38]. In our study, male mice exhibited a significant decline in compliance compared with females. Our data show significantly worse lung function and higher lung collagen deposition in male ACE2 knockouts compared with females. This gender-based difference could suggest a hormonal involvement in the pathophysiology of BLM-induced lung injury. Indeed, 17β-estradiol-mediated upregulation of ACE2 protects the kidney from the progression of hypertensive renal disease and female ACE2 knockout mice showed minimal age-related renal injury [39, 40]. Likewise, other anti-fibrotic agents (i.e., relaxin, an insulin-like hormone secreted during pregnancy with a demonstrated antifibrotic effect in experimentally induced pulmonary fibrosis) may play a role in the decreased collagen deposition seen in females [41]. Recently, Reis et al. reported that ANG [1–7], its receptor Mas, and ACE2 are expressed in the human ovary [42]. This could imply a constant endogenous source of ACE2 that would confer further cardiovascular protection. Besides estrogens, this could provide another explanation for cardiopulmonary protection in females.
Under no disease process, there is evidence that 17β-estradiol exerts differential regulation on components of the RAS, modifying both mostly AT1 receptor genes. In the lung 17β-estradiol administration downregulated AT1 receptor expression. However, no significant interaction in the regulation of AT2 receptor mRNA was evident [43]. Waseda et al. [44] demonstrated that both AT1 and AT2 aid in the pro-fibrotic effect of BLM in the lungs. In our study, lung AT1R expression was unchanged between control and BLM exposed WT female and WT male (Fig. 9a). Conversely, lung AT2R expression was significantly increased in BLM female WT as compared with control female WT and BLM male WT (Fig. 9b). The AT2/AT1 receptor ratio was significantly increased in BLM WT female (Fig. 9c) compared with all other groups. This suggests that a compensatory increase in AT2 could be in part responsible for the decreased fibrosis seen in BLM WT females.
In conclusion, ACE2 exerts a protective effect in BLM-induced lung injury. Human recombinant ACE2 may have therapeutic potential to prevent lung fibrosis after ALI/ARDS. Attenuated fibrosis in BLM exposed ACE2−/− mice compared with ACE2−/y suggest a hormonal involvement in lung fibrosis post ALI/ARDS.
References
Ashbaugh DG, Bigelow DB, Petty TL, Levine BE (1967) Acute respiratory distress in adults. Lancet 2(7511):319–323
Ware LB, Matthay MA (2000) The acute respiratory distress syndrome. N Engl J Med 342(18):1334–1349
Orme J Jr, Romney JS, Hopkins RO, Pope D, Chan KJ, Thomsen G, Crapo RO, Weaver LK (2003) Pulmonary function and health-related quality of life in survivors of acute respiratory distress syndrome. Am J Respir Crit Care Med 167(5):690–694
Miwa C, Koyama S, Watanabe Y, Tsubochi H, Endo S, Nokubi M, Kawabata Y (2010) Pathological findings and pulmonary dysfunction after acute respiratory distress syndrome for 5 years. Intern Med 49(15):1599–1604
Davidson TA, Caldwell ES, Curtis JR, Hudson LD, Steinberg KP (1999) Reduced quality of life in survivors of acute respiratory distress syndrome compared with critically ill control patients. JAMA 281(4):354–360
Briel M, Meade M, Mercat A, Brower RG, Talmor D, Walter SD, Slutsky AS, Pullenayegum E, Zhou Q, Cook D et al (2010) Higher vs lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome: systematic review and meta-analysis. JAMA 303(9):865–873
Cepkova M, Matthay MA (2006) Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. J Intensive Care Med 21(3):119–143
Peter JV, John P, Graham PL, Moran JL, George IA, Bersten A (2008) Corticosteroids in the prevention and treatment of acute respiratory distress syndrome (ARDS) in adults: meta-analysis. BMJ 336(7651):1006–1009
Afshari A, Brok J, Moller AM, Wetterslev J (2010) Aerosolized prostacyclin for acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Cochrane Database Syst Rev (8):CD007733.
Putensen C, Theuerkauf N, Zinserling J, Wrigge H, Pelosi P (2009) Meta-analysis: ventilation strategies and outcomes of the acute respiratory distress syndrome and acute lung injury. Ann Intern Med 151(8):566–576
Rockx B, Baas T, Zornetzer GA, Haagmans B, Sheahan T, Frieman M, Dyer MD, Teal TH, Proll S, van den Brand J et al (2009) Early upregulation of acute respiratory distress syndrome-associated cytokines promotes lethal disease in an aged-mouse model of severe acute respiratory syndrome coronavirus infection. J Virol 83(14):7062–7074
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H et al (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436(7047):112–116
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W et al (2005) A crucial role of angiotensin converting enzyme 2 (ace2) in SARS coronavirus-induced lung injury. Nat Med 11(8):875–879
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC et al (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426(6965):450–454
Marshall RP, McAnulty RJ, Laurent GJ (2000) Angiotensin ii is mitogenic for human lung fibroblasts via activation of the type 1 receptor. Am J Respir Crit Care Med 161(6):1999–2004
Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, McAnulty RJ, Laurent GJ (2004) Angiotensin ii and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol 286(1):L156–164
Wang D, Morales JE, Calame DG, Alcorn JL, Wetsel RA (2010) Transplantation of human embryonic stem cell-derived alveolar epithelial type ii cells abrogates acute lung injury in mice. Mol Ther 18(3):625–634
Xie X, Chen J, Wang X, Zhang F, Liu Y (2006) Age- and gender-related difference of ace2 expression in rat lung. Life Sci 78(19):2166–2171
Casey J, Kaplan J, Atochina-Vasserman EN, Gow AJ, Kadire H, Tomer Y, Fisher JH, Hawgood S, Savani RC, Beers MF (2005) Alveolar surfactant protein d content modulates bleomycin-induced lung injury. Am J Respir Crit Care Med 172(7):869–877
Zhong JC, Ye JY, Jin HY, Yu X, Yu HM, Zhu DL, Gao PJ, Huang DY, Shuster M, Loibner H et al (2011) Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ace2 and profilin-1 expression. Regul Pept 166(1–3):90–97
Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z et al (2010) Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 122(7):717–728, 718 p following 728
Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J, Loibner H, Janzek E, Schuster M, Penninger JM et al (2010) Human recombinant ace2 reduces the progression of diabetic nephropathy. Diabetes 59(2):529–538
Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM et al (2010) Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: Prevention of angiotensin ii-dependent hypertension. Hypertension 55(1):90–98
Zhong J, Guo D, Chen CB, Wang W, Schuster M, Loibner H, Penninger JM, Scholey JW, Kassiri Z, Oudit GY (2011) Prevention of angiotensin ii-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 57(2):314–322
Wong DW, Oudit GY, Reich H, Kassiri Z, Zhou J, Liu QC, Backx PH, Penninger JM, Herzenberg AM, Scholey JW (2007) Loss of angiotensin-converting enzyme-2 (ace2) accelerates diabetic kidney injury. Am J Pathol 171(2):438–451
Kassiri Z, Oudit GY, Sanchez O, Dawood F, Mohammed FF, Nuttall RK, Edwards DR, Liu PP, Backx PH, Khokha R (2005) Combination of tumor necrosis factor-alpha ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase-3 knock-out mice. Circ Res 97(4):380–390
van Haaften T, Byrne R, Bonnet S, Rochefort GY, Akabutu J, Bouchentouf M, Rey-Parra GJ, Galipeau J, Haromy A, Eaton F et al (2009) Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med 180(11):1131–1142
Lovgren AK, Jania LA, Hartney JM, Parsons KK, Audoly LP, Fitzgerald GA, Tilley SL, Koller BH (2006) Cox-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 291(2):L144–156
Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ (1992) Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol 72(1):168–178
Reddy GK, Enwemeka CS (1996) A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem 29(3):225–229
Woessner JF Jr (1961) The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys 93:440–447
Li X, Molina-Molina M, Abdul-Hafez A, Uhal V, Xaubet A, Uhal BD (2008) Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol 295(1):L178–185
Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, Jun JY, Sriramula S, Mariappan N, Pourang D et al (2010) The angiotensin-converting enzyme 2/angiogenesis-(1–7)/mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med 182(8):1065–1072
Han MK, Swigris J, Liu L, Bartholmai B, Murray S, Giardino N, Thompson B, Frederick M, Li D, Schwarz M et al (2010) Gender influences health-related quality of life in ipf. Respir Med 104(5):724–730
Voltz JW, Card JW, Carey MA, Degraff LM, Ferguson CD, Flake GP, Bonner JC, Korach KS, Zeldin DC (2008) Male sex hormones exacerbate lung function impairment after bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 39(1):45–52
Gharaee-Kermani M, Hatano K, Nozaki Y, Phan SH (2005) Gender-based differences in bleomycin-induced pulmonary fibrosis. Am J Pathol 166(6):1593–1606
Kolb M, Bonniaud P, Galt T, Sime PJ, Kelly MM, Margetts PJ, Gauldie J (2002) Differences in the fibrogenic response after transfer of active transforming growth factor-beta1 gene to lungs of “Fibrosis-prone” And “Fibrosis-resistant” Mouse strains. Am J Respir Cell Mol Biol 27(2):141–150
Jegal Y, Kim DS, Shim TS, Lim CM, Do Lee S, Koh Y, Kim WS, Kim WD, Lee JS, Travis WD et al (2005) Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 171(6):639–644
Ji H, Menini S, Zheng W, Pesce C, Wu X, Sandberg K (2008) Role of angiotensin-converting enzyme 2 and angiotensin(1–7) in 17beta-oestradiol regulation of renal pathology in renal wrap hypertension in rats. Exp Physiol 93(5):648–657
Oudit GY, Herzenberg AM, Kassiri Z, Wong D, Reich H, Khokha R, Crackower MA, Backx PH, Penninger JM, Scholey JW (2006) Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin ii-dependent glomerulosclerosis. Am J Pathol 168(6):1808–1820
Tang ML, Samuel CS, Royce SG (2009) Role of relaxin in regulation of fibrosis in the lung. Ann N Y Acad Sci 1160:342–347
Reis FM, Bouissou DR, Pereira VM, Camargos AF, Dos Reis AM, Santos RA (2010) Angiotensin-(1–7), its receptor mas, and the angiotensin-converting enzyme type 2 are expressed in the human ovary. Fertil Steril 95:176–181
Brosnihan KB, Hodgin JB, Smithies O, Maeda N, Gallagher P (2008) Tissue-specific regulation of ace/ace2 and at1/at2 receptor gene expression by oestrogen in apolipoprotein e/oestrogen receptor-alpha knock-out mice. Exp Physiol 93(5):658–664
Waseda Y, Yasui M, Nishizawa Y, Inuzuka K, Takato H, Ichikawa Y, Tagami A, Fujimura M, Nakao S (2008) Angiotensin ii type 2 receptor antagonist reduces bleomycin-induced pulmonary fibrosis in mice. Respir Res 9:43
Acknowledgments
We acknowledge the financial support from the Canadian Institutes of Health Research (GYO grant 86602), Alberta Innovates—Health Solutions (BT and GYO) and the Stollery Children’s Hospital Foundation. GJRP is supported by Alberta Health Innovates. BT is supported by the Alberta Innovates Health Solutions (AIHS), Canada Foundation for Innovation (CFI), and the Canada Research Chairs Program. GYO is a Clinician-Investigator of the Alberta Innovates—Health Solutions and the Distinguish Clinician Scientist of the Heart and Stroke Foundation of Canada and Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rey-Parra, G.J., Vadivel, A., Coltan, L. et al. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. J Mol Med 90, 637–647 (2012). https://doi.org/10.1007/s00109-012-0859-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-012-0859-2