
Abstract
Owing to recent developments in DNA sequencing technologies, whole-genome analysis of crop species has become a routine procedure. In order to improve local rice cultivars adapted to northern Japan, we are applying whole-genome analysis to identify and utilize useful alleles for crossbreeding. Here we show that large-scale generation of genetic resources combined with whole-genome analyses including MutMap and QTL-seq provides a powerful platform for the application of “next-generation breeding” in rice.
Similar content being viewed by others

Keywords
26.1 Introduction
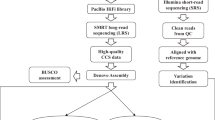
Rice is the principal staple crop in Asia including Japan. The Tohoku region of northern Japan that also includes Iwate Prefecture is the major rice production area in the country. Iwate Biotechnology Research Center (IBRC), in collaboration with Iwate Agricultural Research Center (IARC), has a mandate for basic researches geared toward the improvement of elite local rice cultivars. The main mission of IBRC and IARC is the delivery to local farmers of high-yielding and quality rice cultivars that are competitive on the market, as well as resistant to various biotic and abiotic stresses such as blast disease and too cold summer. To meet this goal, we are employing crossbreeding to improve these elite local rice cultivars. Our approach, which is different from the practices of conventional breeding commonly employed, involves a quick and precise breeding based on genome analysis that targets traits of interest. We call this approach “next-generation breeding.” To this end, we have devoted considerable efforts to (1) establishing large number of rice genetic resources to identify and exploit useful phenotypic traits and (2) developing genome analysis pipelines that enable rapid identification of alleles that confer the desired traits (Fig. 26.1). In this chapter, we would like to share our experiences and achievements from these on-going endeavors.

General scheme of rice “next-generation breeding” (Adopted by IBRC and IARC)
26.2 Rice Cultivar “Hitomebore” Adapted to Northern Japan
The most popular variety currently grown in Iwate is a temperate japonica rice cultivar known as “Hitomebore.” It was developed at Furukawa Agricultural Research Station, Miyagi Prefecture in Tohoku region from a crossbreeding between “Koshihikari,” currently the most widely grown cultivar in Japan, and “Hatsuboshi.” It was registered in 1991 but was widely adopted after 1993 following the severe cold summer that resulted in extremely low yields in rice including “Sasanishiki,” which was the most popular cultivar widely grown in Tohoku region at the time. “Hitomebore” is characterized by strong cold tolerance and excellent eating quality (Table 26.1). These combined with traits that make it suitable for cultivation lead to its wider adoption by farmers over considerable areas in northern Japan. The government census published in 2010 reveals that in terms of total area of production in Japan, “Hitomebore” is second only to “Koshihikari” (http://www.maff.go.jp/j/tokei/sokuhou/syukaku_suitou_09/).
26.3 Whole-Genome Sequence of Rice
The first draft genome sequences of rice were published in 2002 for japonica (Goff et al. 2002) cultivar Nipponbare and indica cultivar 93-11 (Yu et al. 2002), respectively. Following to these, a high-quality whole-genome sequence was published using a japonica cultivar Nipponbare (IRGSP 2005), providing one of the most accurate sequences available for a crop species so far. The “Nipponbare” genome sequence is 389 Mb in size and has set the stage for application of genomics to rice improvement since its release. Useful database and analysis tools are available from Rice Annotation Project Database (RAP-DB) portals (http://rapdb.dna.affrc.go.jp).
26.4 Genetic Resources for Rice Improvement
We have been working over a decade to generate genetic resources that can be utilized for improvement of local rice cultivars. We have generated two categories of resources: mutant lines and recombinant inbred lines (RILs), both in “Hitomebore” genetic background.
26.4.1 Mutant Lines
We have generated a total of 15,000 ethyl methanesulfonate (EMS) mutant lines including 12,000 Hitomebore and 3000 Sasanishiki mutants. EMS is an alkylating chemical that induces primarily GC ➔ AT transition-type nucleotide substitutions. Details of the mutagenesis procedure have been reported elsewhere (Rakshit et al. 2010). Briefly, we treated immature embryos of rice with 0.175% EMS solution immediately after anthesis by immersing flower spikes in a plastic bag containing EMS solution overnight. This method was adopted in order to minimize chimerism, which occurs more frequently in seed mutagenesis. After mutagenesis, matured seeds of the M1 generation were sown, and plants were grown to set flowers, which were self-fertilized to obtain M2 seeds. Ten M2 seeds per M1 line were planted in paddy field to observe their phenotypes. The majority of induced mutations is heterozygous in the M1 generation and brought to homozygous state in the M2 generation. Accordingly, phenotypes of recessive mutations are expected to manifest in one fourth of the M2 individuals. The chance that we obtain recessive homozygous plants among ten M2 individuals is 1−(3/4)10) ≈ 0.94, which allow us to observe majority of the mutant phenotypes in the M2 generation. We re-sequenced “Hitomebore” and identified a total of 128,704 polymorphic sites by aligning “Hitomebore” short reads to the “Nipponbare” reference genome sequence. We then replaced “Nipponbare” nucleotides with “Hitomebore” ones at all the polymorphic sites to generate a “Hitomobore” reference genome. Resequencing of more than 20 independent mutant lines revealed that our mutagenesis on average induced 1500 SNPs per genome per line. This suggested we can expect 46 SNPs per 1000 bp randomly selected genomic region if we screen the entire 12,000 “Hitomebore” mutant lines. This suggested that the majority of genes were targeted by our mutagenesis. These mutant lines have been used for forward genetics as in MutMap analysis (Abe et al. 2012) described below, as well as for TILLING (Till et al. 2003) that represents a reverse genetics approach (Rakshit et al. 2010).
26.4.2 Recombinant Inbred Lines (RILs)
To exploit the natural variation in rice for breeding, we generated RILs using “Hitomebore” as the common parent. RILs are generated by crossing two lines to obtain an F1 progeny, which is then self-fertilized (selfed) to produce F2 progeny. Each F2 individual is treated as a line, and a single F3 seed derived from selfing of F2 is grown to obtain F4 seeds. This process is repeated several times to generate F7–F9 generations (Fig. 26.2). Finally, a large number of seeds are harvested from each line in the advanced generation. RILs offer two major advantages over F2 population in QTL mapping: (1) Majority of the genome is brought to homozygous state in RILs by repeated selfing, meaning the phenotypes are determined by the additive effects of genes with a minimal influence from dominance effects. This increases the precision of QTL mapping. (2) Each F2 genotype is represented only by one individual, whereas each line of RIL has >1000 seeds with practically an identical genotype. This allows the planting of a large number of individuals to perform phenotyping in replicates, resulting in the reduction of phenotypic variance caused by the nongenetic causes. We crossed “Hitomebore” to 22 rice accessions (8 aus, 5 indica, 4 temperate japonica, 5 tropical japonica) presumably representing the O. sativa genetic diversity. Approximately 300 F2 seeds per cross were used as the starting materials for generating RILs by single-seed descent (SSD) method. This generated a total of 3000 RILs of F7–F9 generations, with progeny number in each cross ranging from 30 to 300. These RILs have been planted in IARC paddy field and are being used for QTL-seq (see below) and nested association mapping (NAM) studies (Yu et al. 2008).

A simplified scheme for the generation of recombinant inbred lines (RILs)
26.5 Genome Analysis Methodologies
26.5.1 MutMap
In order to exploit “Hitomebore” mutant lines in rice breeding, we developed MutMap (Abe et al. 2012), which is a whole-genome sequencing (WGS)-based method for the identification of mutations responsible for phenotypes of interest. In MutMap, we cross a mutant of interest to the original line used for the mutagenesis. In our case, a mutant is crossed to a “Hitomebore” wild-type (WT) plant. The resulting F1 is selfed to obtain F2 progeny segregating for the WT and mutant-type progeny in a 3:1 ratio provided that the causative mutation is recessive. This crossing scheme is different from the conventional trait mapping in which a mutant line of interest is crossed to a genetically distant line. The crossing scheme of MutMap allows researchers to target subtle quantitative differences in mutant phenotypes that are difficult to address using distant crosses. DNAs of multiple mutant F2 progeny (usually 20 individuals) are pooled to make a DNA bulk, which is subjected to WGS by the illumina sequencing platform. The resulting short sequence reads of 100–150 bp are aligned to the reference sequence of “Hitomebore.” Among the population of F2 progeny showing the mutant phenotype, the frequency of mutated allele at the causative gene is 100%, whereas that of mutations in the genomic regions not related to the mutant phenotype is expected to be 50%. The alignment of short reads obtained from bulked DNA to the reference genome sequence reveals the allele frequencies of all SNPs across the genome (Fig. 26.3). When short reads are aligned to the reference genome, each nucleotide position is usually covered by multiple reads. Here we introduce a measure named SNP-index. Within ten short reads that are aligned to a given genomic region, if all reads had a SNP at a particular nucleotide different from the reference nucleotide, the SNP-index is 10/10 = 1.0. On the other hand, if four of the reads have SNPs and the rest have the nucleotide identical to the reference nucleotide, SNP-index is 4/10 = 0.4. This SNP-index is in fact the frequency of particular SNPs among the population of pooled individuals. Next, we plot a graph relating genomic position of an SNP on the x-axis and its SNP-index value on the y-axis. As explained above, the SNP-index value of the causative SNP responsible for the mutant phenotype should be 1, and those of SNPs on the chromosomes that do not contain the causative SNP are 0.5. However, SNP-index values of the SNPs tightly linked to the causative SNP show higher SNP-index values (0.5< SNP-index <1) due to linkage drag. Therefore, the graph typically shows a cluster of SNPs with higher SNP-index values near the causative one, allowing a rapid identification of causative mutation. The result of a typical MutMap analysis is given in Fig. 26.4. As extensions of the MutMap methodology, we have developed MutMap-Gap (Takagi et al. 2013a) and MutMap+ (Fekih et al. 2013).

Alignment of short reads to reference genome sequence and definition of SNP-index values

SNP-index plot of the rice 12 chromosomes generated by MutMap analysis. A candidate region in chromosome 12 is indicated by red triangle. Blue dot, SNP; red line, sliding window average of SNP-index; and green and orange lines, sliding window average of 95%- and 99%-confidence level, respectively, under the null hypothesis
26.5.2 QTL-Seq
To identify quantitative trait loci (QTL) controlling phenotypic differences between two cultivars, we developed a WGS-based method named QTL-seq (Takagi et al. 2013b). This method is suitable for identification of QTL responsible for natural variation of phenotypes. In QTL-seq, two cultivars are crossed and segregation of phenotypes is observed either in F2 progeny or RILs. The majority of quantitative traits are controlled by multiple genes. Accordingly, the frequency of phenotypes among F2 progeny or RILs typically follows the normal distribution. We then focus on the progeny with extreme phenotypes, those with the minimum and maximum phenotypic values. DNAs of 10–20 progeny with the minimum values are pooled to make the low (L) bulk, and those with the maximum values are pooled to make the high (H) bulk. The L and H bulk DNAs are separately subjected to WGS, and the resulting short reads are aligned to the reference genome sequence generated for either of the parents used for crossing. For each SNP present between the two parents, we obtain SNP-index values and generate a graph relating SNP position and SNP-index as explained above in MutMap. If we use the genome sequence of the parent 1 as the reference, alignment of short reads only derived from the parent 1 genome gives SNP-index value = 0, whereas alignment of reads only from the parent 2 genome gives SNP-index value = 1. Genomic regions not related to the selection of phenotype between L and H are expected to be transmitted equally from the two parents, bringing the SNP-index values to 0.5. The genomic regions responsible for the L and H separation of phenotypes should have SNP-index that significantly deviates from 0.5. Typically, we observed a peak of SNP-index for either of the two bulks (i.e., L bulk) and a valley of SNP-index in the other bulk (H bulk) at the identical genomic position. This identifies the location of QTL controlling the difference of phenotype between the two parental cultivars. A typical result of QTL-seq analysis is shown in Fig. 26.5.

QTL-seq result. These graphs show SNP positions (x-axis) and delta-SNP-Index values (y-axis) deplcting the difference in SNP-Index values between the H- and L-bulks. Three candidate QTLs are indicated by red triangles. Blue dot, SNP; red line, sliding window average of SNP-index; and green and orange lines, sliding window average of 95%- and 99%-confidence intervals, respectively, under the null hypothesis of no QTL
26.6 Examples of Next-Generation Breeding of Rice
26.6.1 Salt-Tolerant Cultivar “Kaijin”
Following the devastating 2011 earthquake and tsunami that attacked Tohoku area of Japan, >20,000 ha of rice paddy field facing the Pacific coast was inundated with seawater, resulting in salt contamination of agricultural land. As the local rice cultivar “Hitomebore” is not tolerant to salt stress, we set out to develop a salt-tolerant rice cultivar in “Hitomebore” genetic background. Accordingly, we carried out a genetic screen for salt tolerance using 6000 EMS mutant lines of “Hitomebore” and identified a mutant that survived 1.5% NaCl supplied to the soil with irrigation water for 7 days. This candidate mutant line was designated as hitomebore salt tolerant 1 (hst1).
To identify the mutation responsible for the high salinity tolerance of hst1, we applied MutMap. F2 progeny derived from a cross between hst1 and “Hitomebore” WT was treated with water containing 0.75% NaCl. The progeny segregated in a 133:54 ratio for salinity-susceptible and salinity-tolerant phenotypes, respectively, conforming to a 3:1 segregation ratio that indicated the salinity tolerance of hst1 is conferred by a single recessive mutation. DNA was pooled from 20 individuals that showed salinity tolerance and applied to whole-genome resequencing using illumina DNA sequencer. The causative SNP at the nucleotide position 4,138,223 on chromosome 6 corresponded to the third exon of the Os06g0183100 gene, which is predicted to encode a B-type response regulator designated as OsRR22. We only required a year to identify the causative SNP conferring the high salinity tolerance of hst1 by MutMap.
To develop a salinity-tolerant cultivar that has the hst1 mutation, we backcrossed “Hitomebore” to hst1. After two backcrossing events followed by two consecutive selfings (BC1F3), combined with confirmation of inheritance of the recessive hst1 allele by Sanger sequencing, we developed a line named “Kaijin” (Neptune in Japanese). “Kaijin” has the same level of salt tolerance as hst1, and whole-genome resequencing revealed that “Kaijin” differed from Hitomebore WT by only 201 SNPs, which is a significant reduction from the 1088 homozygous SNPs in the hst1. “Kaijin” is practically equivalent to the elite cultivar “Hitomebore” in all agronomic traits except for the salinity tolerance (Takagi et al. 2015).
The example described above has demonstrated the fact that screening of mutant lines combined with MutMap allows the accelerated breeding of cultivar with desirable traits. This is a powerful tool that is now at the disposal of breeders, allowing them to respond to immediate and pressing demands such as the ones caused by global climate changes or natural as well as mandate disasters.
26.6.2 Good Eating Quality Cultivar “Konjiki-no-kaze”
Eating quality of rice determines consumers’ preferences and market price. Non-glutinous temperate japonica rice cultivars are consumed as staple in Japan, where soft and slightly sticky rice is generally preferred. Amylose content of rice grain’s starch is an important factor that affects the eating quality. Therefore, fine-tuning of amylose content is one of the most important targets in rice breeding program in Japan. The gene encoding granulebound starch synthase I enzyme (GBSSI), named Waxy (Wx), is required for amylose synthesis in rice. Several Wx mutants have been used to breed rice lines with low amylose content, resulting in the release of several cultivars (e.g., “Milky Queen,” “Yumepirika”). However, these Wx mutant alleles are not suitable as genetic resources to use in Tohoku region because these alleles make grain amylose much lower than the optimal range (14–17% amylose content) that is generally preferred by consumers.
To identify a novel genetic resource of low amylose content suitable for Tohoku region, we carried out a screen of 1600 EMS mutant lines of “Hitomebore” and identified a mutant line “Hit1073” with amylose content lower by only 2.5% compared to “Hitomebore” (17–19%). To identify the causative mutation responsible for the slightly low amylose content, we applied MutMap and succeeded in detecting a few candidate SNPs associated with low amylose content in a genomic region different from that of Wx. Using one of these SNPs as a DNA marker, we performed marker-assisted selection (MAS) to the F2 progeny derived from the cross between Hit1073 and Hitomebore WT. Finally, we developed a new cultivar named “Konjiki-no-kaze” (gold-colored wind). This cultivar received a high rating in eating quality that has resulted from slightly low amylose content. We expect this cultivar to be widely planted by farmers in Iwate Prefecture in the near future.
26.7 Prospects
Rapid progresses in DNA sequencing technologies have enabled efficient genome analysis of crops (Varshney et al. 2014 ). Combined with suitable genetic resources and good phenotyping protocols, breeders are now capable of implementing crop improvement with a high precision in a short time. We hope that the experiences accumulated in rice improvement would be transferred to other crop species to allow high-speed breeding of crops that can adapt to rapid environmental changes as well as contribute to global food security.
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H et al (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Fekih R, Takagi H, Tamiru M, Abe A, Natsume S et al (2013) MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS One 10:e68529
Goff SA, Ricker D, Lan T-H, Presting G, Wang R et al (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296:92–100
IRGSP (2005) The map based sequence of the rice genome. Nature 436:793–800
Rakshit S, Kanzaki H, Matsumura H, Rakhist A, Fujibe H et al (2010) Use of TILLING for reverse and forward genetics of rice. In: Kahl G, Meksem K (eds) The handbook of plant mutation screening. Wiley-VCH, Weinheim
Takagi H, Uemura A, Yaegashi H, Tamiru M, Abe A et al (2013a) MutMap-gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol 200:276–283
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S et al (2013b) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Takagi H, Tamiru M, Abe A, Yoshida K, Uemura A et al (2015) MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat Biotechnol 33:445–449
Till BJ, Stevene HR, Greene EA, Comodo CA, Enns LC et al (2003) Large-scale discovery of induced point mutations with high-throughput TILLING. Genome Res 13:524–530
Varshney R, Terauchi R, McCouch S (2014) Harvesting the promising fruits of genomics: applying genome sequencing technologies to crop breeding. PLoS Biol 12(6):e1001883. https://doi.org/10.1371/journal.pbio.1001883
Yu J, Hu S, Wang J, Wong GKS, Li S (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296:79–92
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551
Acknowledgments
This study was supported by the grant from “Science and technology research promotion program for agriculture, forestry, fisheries and food industry.” We extend our gratitude to Dr. S. kuroda for a general support and Prof. T. Sasaki and Prof. M. Ashikari for improvement of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Abe, A. et al. (2018). Next-Generation Breeding of Rice by Whole-Genome Approaches. In: Sasaki, T., Ashikari, M. (eds) Rice Genomics, Genetics and Breeding. Springer, Singapore. https://doi.org/10.1007/978-981-10-7461-5_26
Download citation
DOI: https://doi.org/10.1007/978-981-10-7461-5_26
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7460-8
Online ISBN: 978-981-10-7461-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)