
Abstract
Oxazolidinones are a class of synthetic antimicrobial agents with potent activity against a wide range of multidrug-resistant Gram-positive pathogens including methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci. Oxazolidinones exhibit their antibacterial effects by inhibiting protein synthesis acting on the ribosomal 50S subunit of the bacteria and thus preventing formation of a functional 70S initiation complex. Currently, two oxazolidinones have been approved by the US Food and Drug Administration: linezolid and more recently tedizolid. Other oxazolidinones are currently under investigation in clinical trials. These antimicrobial agents exhibit a favourable pharmacokinetic profile with an excellent bioavailability and a good tissue and organ penetration. In-vitro susceptibility studies have shown that oxazolidinones are bacteriostatic against enterococci and staphylococci, and bactericidal for the majority of strains of streptococci. In the context of emergence of resistance to glycopeptides, oxazolidinones have become an effective alternative to vancomycin treatment frequently associated with nephrotoxicity. However, oxazolidinones, and linezolid in particular, are associated with significant adverse events, myelosuppression representing the main unfavourable side effect. More recently, tedizolid has been shown to effectively treat acute bacterial skin and skin structure infections. This newer oxazolidinone offers the advantages of once-daily dosing and a better safety profile in healthy volunteer studies (fewer gastrointestinal and haematological side effects). The potential use of tedizolid for other infections that could require longer therapy warrants further studies for positioning this new oxazolidinone in the available antimicrobial armamentarium. Moreover, other oxazolidinones are currently under active investigation.
Similar content being viewed by others

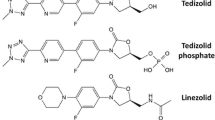
Oxazolidinones are a class of synthetic antibiotics with an activity against Gram-positive pathogens including methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci, approved for the treatment of acute bacterial skin and skin structure infections (linezolid and tedizolid), community-acquired and nosocomial pneumonia (linezolid). |
Tedizolid is a new oxazolidinone exhibiting a favourable pharmacokinetic and safety profile and potential activity against linezolid-resistant strains. |
Further research is needed to determine the place of tedizolid in the antimicrobial arsenal. |
1 Introduction
Oxazolidinones represent a class of antimicrobial agents with a strong activity against Gram-positive bacteria. They were firstly discovered in the late 1980s but their development was discontinued because of safety issues involving hepatotoxicity [1]. However, as the emergence of resistance to glycopeptides has become a major concern for the hospital infection community, especially in methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE), interest returned to oxazolidinones in the 1990s. Consequently, linezolid was the first oxazolidinone approved and released by the US Food and Drug Administration in April 2000. A decade elapsed before tedizolid, the second oxazolidinone was approved by the Food and Drug Administration in 2014 and by the European Medicine Agency in 2015 [2, 3].
Oxazolidinones act as a protein synthesis inhibitor on the ribosomal 50S subunit of the bacteria targeting the initiation phase of the protein synthesis. This leads to functional initiation and messenger RNA translation blockage. This mechanism of action differs from other protein synthesis inhibitors such as chloramphenicol, tetracyclines, lincosamides or macrolides. This original mechanism of action prevents oxazolidinones from cross-resistance to pathogens harbouring genes that affect the ribosome, rendering antimicrobials such as lincosamides and macrolides ineffective.
Oxazolidinones exhibit a good activity against the vast majority of Gram-positive microorganisms including less susceptible pathogens such as VRE and MRSA [4]. These antimicrobial anti-infective agents have become an effective alternative to vancomycin treatment frequently associated with nephrotoxicity and emergence of resistance to glycopeptides. Linezolid has also demonstrated potent in vitro activity against Mycobacterium tuberculosis, Corynebacterium spp., Moraxella catarrhalis, Nocardia spp., Listeria monocytogenes and Bacteroides fragilis [5, 6]. This report reviews the available data on the pharmacokinetics (PK)/pharmacodynamics and toxicodynamics of the two approved oxazolidinones, linezolid and tedizolid as well as their main clinical indications.
2 Methods
A comprehensive literature search was conducted using PubMed and Web of Science for all materials containing the names: [linezolid OR tedizolid OR tedizolid phosphate OR torezolid OR oxazolidinones] AND [pharmacokinetics OR pharmacodynamics OR PK OR PD OR clinical indications]. The search included citations published up to May 2017. From the retrieved citations, only those fulfilling the following criteria were included in this review: original research article, clinical and experimental studies reporting at least one pharmacokinetic or pharmacodynamic parameter or clinical outcome as study outcome.
3 Linezolid
3.1 Linezolid Pharmacokinetics
3.1.1 Pharmacokinetic Profile
The pharmacokinetic profile of linezolid has been largely studied in healthy volunteers [7,8,9,10] (Table 1). Linezolid is an amphiphilic drug exhibiting a time-dependent activity against mainly Gram-positive pathogens. Linezolid also has limited concentration-dependent killing activity and modest post-antibiotic effects. The plasma protein binding of linezolid is approximately 31% (range 4–32%) and is concentration independent [11, 12]. The volume of distribution of linezolid at steady state averaged 36.1–47.3 L in healthy adult volunteers [10, 13]. Animal and human pharmacokinetic studies have demonstrated that linezolid readily distributes to well-perfused tissues [14]. Linezolid is rapidly and completely absorbed after oral administration, with absolute bioavailability of 100%, thus allowing early switch from intravenous to oral administration [15]. Linezolid is 30% renally excreted as unchanged drug whereas 50% of linezolid is primarily metabolised by oxidation of the morpholine ring into two inactive carboxylic acid metabolites [10]. The elimination half-life varies from 3 to 7 h, requiring twice-daily dosing regimens [9]. A small degree of nonlinearity has been observed, with a 30% decrease in clearance after a fivefold increase in dose. The nonlinearity seemed not relevant over the therapeutic dosage range [10]. Nevertheless, a population pharmacokinetic analysis, from a large cohort of patients treated in a compassionate use programme, has highlighted that a parallel-first order plus Michaelis–Menten model best described the data suggesting some degree of non-linearity [16]. In this population pharmacokinetic analysis, ideal body weight and creatinine clearance both affected linezolid PK.
3.1.2 Linezolid Tissue Penetration
Antibiotic tissue penetration and distribution depend on physicochemical properties of substances and pathophysiological changes in patients.
3.1.2.1 Intrapulmonary Penetration
Linezolid is widely used to treat community acquired and nosocomial pneumonia owing to the emergence of MRSA pneumonia, especially in USA [17,18,19,20]. The pharmacokinetic properties of linezolid are of interest in pneumonia as its distribution in epithelial lining fluid (ELF) is almost 100% [21, 22]. In a piglet model of MRSA pneumonia, linezolid concentrations in ELF and lungs were higher than linezolid concentrations in blood [23]. In a study including critically ill patients, the mean ± standard deviation linezolid peak and trough concentrations were 17.7 ± 4.0 and 2.4 ± 1.2 mg/L in plasma and 14.4 ± 5.6 and 2.6 ± 1.7 mg/L in ELF, respectively [21]. High pulmonary penetration has also been reported in obese patients especially when continuous infusion of linezolid is used [24].
3.1.2.2 Cerebrospinal Fluid Penetration
As linezolid is bacteriostatic and not bactericidal against S. aureus, it was not initially considered as an antimicrobial candidate for central nervous system (CNS) infections. However, linezolid has been reported to have a favourable pharmacokinetic profile exhibiting a large volume of distribution and a relatively low protein binding, favouring its penetration into the CNS with an area under the concentration–time curve (AUC)CSF/AUCplasma close to 1 [25]. Animal models of MRSA meningitis have confirmed the good penetration of linezolid through the blood–brain barrier and an effective bacterial clearance from cerebrospinal fluid (CSF) at 24 h of treatment [26, 27]. In humans, studies in post-neurosurgical infections have reported a ratio between CSF and plasma linezolid trough concentrations between 0.66 and 1, resulting in maximum concentrations of up to 7.1 ± 2.2 mg/L in CSF at steady state after intravenous infusion of 600 mg every 12 h [28,29,30,31]. Moreover, the half-life of linezolid has been reported as prolonged in CSF (6.8 ± 6 h) compared with plasma (3.7 ± 1 h) [29]. These findings emphasise that linezolid may achieve adequate CSF concentrations in patients requiring antimicrobial therapy for CNS infections caused by Gram-positive pathogens.
3.1.2.3 Skin and Soft-Tissue Penetration
Hospital and community-acquired acute bacterial skin and skin structure infections (ABSSSIs) due to multidrug resistant (MDR) Gram-positive pathogens (MRSA, VRE, vancomycin intermediate-resistant S. aureus, heterogeneous vancomycin intermediate resistant S. aureus and glycopeptide intermediate-resistant S. aureus) are of particular concern. In healthy volunteers, linezolid concentrations in soft tissues are considered sufficient to inhibit growth of several relevant bacteria [14]. After multiple doses, the mean (± standard deviation) ratio of AUC from 0 to 8 h (AUC0–8) to the AUC0–8 for free plasma were 1.4 ± 0.3 and 1.3 ± 0.4 for adipose and muscle tissues, respectively [14]. However, in critically ill and diabetic patients, antimicrobial tissue penetration can be altered [32]. Clinical studies in diabetic foot infections and soft-tissue infections have shown a mean AUC0–12 h value of 114.1 ± 31.8 mg·h/L, a mean minimum plasma concentration (C min) of 5.4 ± 2.4 mg/L and a good penetration into inflamed diabetic foot infected tissues with tissue/plasma ratios of mean 101.7% (95% confidence interval 55.9–147.6) [33]. In nine diabetic patients with chronic lower limb infections, the mean areas under the concentration–time curves from 0 to 12 h for the free fraction of linezolid (∫AUC0–12 values) ± standard deviation for plasma, wound tissue and thigh tissue were 51.24 ± 12.72, 82.76 ± 59.01 and 92.52 ± 60.44 mg·h/L, respectively. Tissue penetration ratios (tissue ∫AUC to plasma ∫AUC) were similar for thigh (1.42; range 1.08–2.23) and wound (1.27; range 0.86–2.26) tissues (p = 0.648) [11]. In critically ill patients, the penetration of linezolid into interstitial tissues exhibits a high inter-individual variability ranging from 20.2 to 144% (mean value: 80%) [34] without influence of sepsis severity [35]. Based on these pharmacokinetic findings, linezolid appears to be a good alternative for ABSSSIs when difficult-to-treat Gram-positive pathogens are suspected.
3.1.2.4 Bone Penetration
Bone composition differs from other tissues and it is difficult to predict whether antimicrobial agents exhibiting a good penetration into other tissues will also achieve adequate concentrations in bone. Linezolid bone penetration is controversial [36, 37]. In 12 patients undergoing hip replacement surgery, the mean ratio of bone-to-serum linezolid concentrations was 0.51 (95% confidence interval 0.43–0.75) [36]. In a clinical study using microdialysis to measure bone concentrations in diabetic foot infections, linezolid tissue penetration assessed by the ratio of free AUC0–12 (∫AUC0–12) in bone to the ∫AUC0–12 in plasma was higher (1.09 ± 0.11) [37]. Penetration of linezolid into bone and joint tissues was studied in 13 patients experiencing implant-associated infections with MRSA [38]. Mean concentrations of linezolid in infected tissues around the prosthesis were greater than 10 mg/L, except in bone specimens, where they reached 3.9 ± 2.0 mg/L [38]. Penetration of linezolid into osteo-articular tissue and fluid was studied in ten patients undergoing primary total knee replacement. Linezolid 600 mg 12 hourly was given orally over the 48 h before the operation and intravenously 1 h before the induction of anaesthesia. Mean concentrations of linezolid at 90 min after the final dose, in serum, synovial fluid, synovium, muscle and cancellous bone, were at least twice the MIC90 for staphylococci and streptococci [39]. After a single dose of 600 mg linezolid in patients undergoing routine cardiopulmonary bypass surgery, penetration of linezolid into the body of the sternum was 44.28% (range 37.76–49.87%) at 2 h and 31.93% (range 17.06–53.35%) at 5 h [40]. Small observational studies have shown a favourable outcome in patients treated with prolonged linezolid therapy for osteomyelitis and/or prosthetic infections but have also reported a high incidence of adverse events (AEs) [41,42,43,44].
3.1.2.5 Heart Valve Penetration
Although bactericidal antimicrobial agents represent the first choice for endocarditis treatment, the emergence of MDR Gram-positive bacteria led to the reconsideration of bacteriostatic agents such as linezolid. However, few clinical data are available to support the use of linezolid in MRSA and vancomycin intermediate-resistant S. aureus endocarditis treatment [45]. In a review of 23 case reports and three case series, Falagas et al. have reported a clinical cure rate of 63.6% after linezolid therapy [45]. Experimental data from animal endocarditis models do not support the superiority of linezolid over vancomycin [46, 47]. Treatment with linezolid induced a mean reduction in aortic valve vegetation bacterial counts of 2.44 log10 colony forming units/g (p < 0.05 compared with controls). However, no linezolid concentrations in valve vegetation were measured in this study. Further randomised clinical trials are needed to determine whether linezolid could represent an alternative option in the treatment of infective endocarditis.
3.1.2.6 Biofilm and Antitoxin Properties
Linezolid had been reported to reduce S. aureus virulence factor expression in vitro [48]. Together with a good intrapulmonary penetration, antitoxin properties of linezolid could represent an advantage for treating toxin-producing S. aureus. Animal models of necrotizing MRSA pneumonia have shown suppressed bacterial production of Panton–Valentine leukocidin and α-hemolysin, reduced production of the neutrophil-chemoattractant interleukin-8 in the lungs and improved survival outcomes in the group treated with linezolid [49]. In an open-label clinical trial comparing linezolid with vancomycin, for nosocomial pneumonia treatment, clinical and microbiological cure rates were equivalent [50, 51]. However, in post-hoc analyses from these trials, linezolid clinical response was superior to vancomycin for hospital-associated MRSA pneumonia [52]. These findings have been lately confirmed in a randomised controlled trial [53]. The effects of linezolid on biofilm inhibition or growth remain to be further evaluated [54].
3.1.3 Linezolid Pharmacodynamics
Linezolid exhibits minimal bactericidal activity against staphylococci, high bactericidal activity against streptococci and is bacteriostatic against Gram-positive cocci such as enterococci. The time plasma drug concentrations remain above the minimal inhibitory concentration (%T > MIC) and the AUC above the minimal inhibitory concentration (AUC/MIC) are the (pharmacokinetic/pharmacodynamic) indexes that best correlate with linezolid efficacy [7, 10, 55]. In a mouse thigh infection model, the AUC/MIC ratio was the most important pharmacokinetic/pharmacodynamic index for linezolid against strains of S. aureus and Streptococcus pneumoniae; however, the %T > MIC was also an important index to consider especially for staphylococci [55]. The pharmacodynamic analysis from a compassionate use programme has shown a linear relationship between %T > MIC and AUC/MIC at AUC/MIC values less than 120. Beyond this value, the correlation between these two pharmacokinetic/pharmacodynamic indexes no longer exists [56]. Higher success rates may occur at AUC/MIC values of 80–120 for bacteraemia and ABSSSIs [56]. Clinical success rates also appear to be higher when concentrations remain above the MIC for the entire interval [56]. Some recent studies have reported inadequate linezolid trough concentrations ( < 2 or > 10 mg/L) in 30–40% patients receiving conventional dosing regimens, leading to inadequate drug exposure and potential toxicity [57, 58]. However, the small sample size of these studies did not allow any correlation with poor clinical outcomes.
3.1.4 Linezolid Dosing Considerations and Therapeutic Drug Monitoring
Experimental data from a rabbit S. aureus endocarditis model have demonstrated that continuous infusion of linezolid was associated with in-vivo linezolid bactericidal activity, suggesting that continuous infusion of linezolid may be an alternative mode of administration in severe infections [47]. In ventilator-acquired pneumonia, the alveolar diffusion of linezolid administered by continuous infusion was 100% and the plasma and ELF concentrations remain above the susceptibility breakpoint for S. aureus (4 mg/L for the entire dosing interval) [22]. Comparing intermittent to continuous infusion of linezolid, Adembri et al. reported better pharmacokinetic/pharmacodynamic target attainment in the continuous infusion group especially when the highest MICs were considered (MIC between 2 and 4 mg/L) [59].
High doses of linezolid, i.e. 600 mg 8 hourly, have also been studied aiming to improve the probability of pharmacokinetic/pharmacodynamic target attainment [60]. In a small cohort of critically ill patients, clinical cure was achieved in 7/7 (100%) of patients. However, 57.1% of patients manifested haematological toxicity associated with high trough concentrations. The concomitant use of therapeutic drug monitoring (TDM) could help optimise drug exposure in difficult-to-treat infections and at-risk populations such as critically ill patients. In a retrospective study including 45 patients undergoing linezolid TDM, Pea et al. has emphasised the need for linezolid TDM in about 30% of studied patients to avoid either drug overexposure leading to toxicity or sub-therapeutic concentrations leading to clinical failure [58].
More recently, the same authors confirmed in a larger cohort of 1049 patients over 10 years that routine linezolid TDM aiming to maintain trough concentrations within a desirable range of 2–7 mg/L may be a valuable tool to optimise drug exposure and to reduce toxicity given the wide inter-individual variability observed in trough concentrations [61, 62]. The good linear relationship between C min and estimated AUC0–24 (r 2 = 0.85) suggests that C min may represent a useful predictor of linezolid total body exposure in daily clinical practice [11]. As the MIC90 of linezolid against staphylococci and enterococci is 2 mg/L [63], Pea et al. have shown that the achievable AUC0–4 in the presence of C min of > 2 mg/L is > 160 mg·h/L, meaning that for a C min of linezolid > 2 mg/L both the pharmacodynamic targets of efficacy (C min higher than MIC90 and AUC/MIC90 ratio of > 80) may be attained [63]. A toxicodynamic model has shown that a C min of > 8.06 mg/L may result in thrombocytopenia by inhibiting platelet synthesis by 50% [64]. Matsumoto et al. also redefined this threshold of 8 mg/L to minimise linezolid-induced thrombocytopenia [65]. Thus, we suggest maintaining linezolid trough concentrations within a range of 2–8 mg/L to ensure optimal drug exposure.
3.1.5 Resistance to Linezolid
Although early linezolid-resistant strains have been reported after linezolid release, results from post-Food and Drug Administration approval surveillance study groups have shown the rate of resistance to linezolid development to be relatively low and stable since its clinical approval [66]. The two most common resistance to linezolid mechanisms are mutations to the 23S ribosomal RNA and the presence of a transmissible chloramphenicol-florfenicol resistance (cfr) ribosomal methyltransferase [67, 68]. Acquisition of the cfr gene results in resistance to linezolid, as well as other antimicrobials such as phenicols, lincosamides, streptogramin A and 16-member-ring macrolide antibiotics. The potential for horizontal transmission of this mechanism of resistance potentially leading to widespread dissemination is a major concern.
3.1.6 Linezolid Safety Profile and Adverse Events
Adverse events have been mainly described with linezolid, which has been widely used over the last 17 years. The main reported side effects range from moderate (asthenia, vomiting, weight loss) to severe AEs: anaemia, thrombocytopenia, peripheral and/or optical neuropathy and severe lactic acidosis [69, 70]. These AEs have been mainly reported in prolonged treatment. For a short treatment duration, severe AEs are rare and oxazolidinones have an interesting safety profile when comparing with vancomycin, which remains the reference drug.
3.1.6.1 Haematological Toxicity
Myelosuppression has been reported in 1 over 750 patients. The mechanisms of myelosuppression are not clearly elucidated, even if inhibition of mitochondria function has been widely evoked, though not proven [64, 70, 71]. Recent population pharmacokinetic and toxicodynamic models have highlighted that inhibition of platelet formation was the main mechanism of action to explain linezolid-induced thrombocytopenia [64, 72]. The toxicity of linezolid is dose dependent [73, 74]. The co-administration of rifampicin and linezolid could decrease the myelosuppression risk [75]. Thrombocytopenia is the main side effect of linezolid. In post-marketing studies, thrombocytopenia has been reported in 1 over 1700 patients [76]. In phase III studies, thrombocytopenia has been documented in 2–4% of patients, only when treatment duration exceeded 14 days [77, 78].
While treatment duration is a key issue for linezolid-induced thrombocytopenia, renal failure is the second major factor [75]. During renal dysfunction, linezolid blood concentrations have been shown to increase and to be correlated with thrombocytopenia risk, in a dose-dependent manner [73]. Combining renal function assessment and trough linezolid concentration monitoring helps avoid myelosuppression [65]. It is therefore recommended to monitor platelet counts once a week in the case of renal failure or treatment duration exceeding 14 days [70]. The risk significantly increases at day 10, whereas therapy for 5–7 days is considered as safe [64]. The risk of thrombocytopenia also depends on the basal platelet count. The risk increases when the basal platelet count is below 150 × 109/L [64]. Stopping treatment is frequently associated with resolution of thrombocytopenia [70].
Anaemia is the second haematological side effect. It occurs in 1 in 2900 exposed patients [76]. As for thrombocytopenia, anaemia increases with treatment duration and renal dysfunction [73, 76].
3.1.6.2 Neurological Toxicity
Peripheral and optic neuropathy has been described with prolonged use of linezolid (12–15 months), mainly during treatment of mycobacterial infection [79,80,81,82]. This complication occurs after a median of 5 months [83]. Peripheral neuropathies are generally sensory, with paraesthesia, numbness and peripheral pain. Electromyography shows severe sensory-motor axonal neuropathy [80, 83]. The prognosis of peripheral neuropathy is poor, with a high rate of non-recovery. Optic neuropathy is generally less severe, with complete or partial recovery in the majority of cases over a few weeks after stopping treatment [83]. If blurred vision is observed in 30% of patients, the occurrence of documented optic neuropathy ranges from 1 to 13% of patients with prolonged treatment [82, 84]. In this context, a regular ophthalmologic screening is mandatory.
3.1.6.3 Lactic Acidosis
Lactic acidosis is a rare but potentially life-threatening complication of linezolid administration. Linezolid inhibits bacterial growth by binding to bacterial ribosomes (16S subunit) and therefore blocking bacterial protein synthesis [85]. Some mitochondrial DNA polymorphisms may induce cross-reaction between linezolid and human mitochondrial ribosomes with secondary inhibition of human mitochondrial activity [71, 86, 87]. In extreme cases, mitochondrial toxicity can cause whole-body energetic failure, with decline of oxygen extraction, vasodilatory shock, acute respiratory and renal failure, leading to rapid fatal issue [88].
The diagnosis of such an AE can be very difficult to distinguish from spontaneous evolution of septic shock. It is therefore important to consider this potential diagnosis whether secondary aggravation of septic shock occurs after initial stabilization. Muscle biopsy can help to diagnose this complication, showing diminished activity and expression of respiratory chain subunits encoded by mitochondrial DNA. However, such analysis remains only possible in highly specialised laboratories [88]. Therefore, unexpected mental confusion, acute respiratory failure and shock occurring during linezolid treatment should lead to the investigation of potential linezolid-induced lactic acidosis. Early diagnosis and prompt linezolid discontinuation have been associated with total resolution of symptoms and favourable outcomes [89]. The onset of linezolid-related lactic acidosis is usually delayed ( > 6 weeks) [89,90,91], but some rapid onset (few days) has been reported in the literature [88, 92]. Therefore, monitoring of lactate levels during linezolid treatment is probably useful.
3.1.6.4 Other Toxic Events
Some cases of pancreatitis and hypoglycaemia had also been reported [92]. Hypoglycaemia can be related to monoamine oxidase (MAO) inhibition and dopaminergic properties [92]. Such mechanisms also imply a caution with the use of linezolid in hypertensive patients or in co-administration with serotoninergic or adrenergic agents [77].
3.1.7 Linezolid Drug Interactions
Linezolid was first studied as a potential reversible inhibitor of a MAO antidepressant. As mentioned above, co-administration of linezolid with other inhibitors of MAO can worsen a serotonin syndrome. As cytochrome P450 enzymes do not play a significant role in linezolid metabolism, major pharmacokinetic drug–drug interactions with these drugs are not expected. Nevertheless, few case reports have demonstrated interactions between rifampicin, levothyroxine or warfarin leading to drug underexposure [93,94,95].
3.1.8 Linezolid Clinical Indications
Linezolid is indicated for suspected or documented infection to penicillin-resistant Gram-positive bacterial infections, especially MRSA (Table 2). This antibiotic is considered as a promising alternative to glycopeptides, which remain the reference therapeutic class for such infections. In this section, we review the potential clinical interest of linezolid over glycopeptides. Considering its excellent distribution into the interstitial subcutaneous fluid (ISF) and lung, linezolid is particularly effective for the treatment of pneumonia and ABSSSIs. Linezolid has also been considered for the treatment of nosocomial meningitis and bone infection. Its role during bloodstream infections (BSI) and endocarditis remains controversial. Finally, linezolid represents an important adjunct to treatment of resistant mycobacteria infection. This specific issue will not be discussed in this article.
3.1.8.1 Theoretical Clinical Advantages of Linezolid
Linezolid offers some advantages that could lead physicians to consider oxazolidinones in Gram-positive infections over glycopeptides in daily practice: available oral route; no venous toxicity requiring central venous access; no renal toxicity; and activity against glycopeptide-resistant Gram-positive bacteria.
3.1.8.2 Pneumonia
Despite an excellent pulmonary diffusion of linezolid, the superiority of linezolid over vancomycin (the reference drug) for MRSA pneumonia treatment in terms of mortality has not been demonstrated both in intensive care unit (ICU) and non-critically-ill patients [96,97,98,99]. Nevertheless, linezolid is associated with a better microbiological eradication, a better clinical success and a significant reduction in acute renal failure as compared with vancomycin [99,100,101]. This leads to a significant reduction in hospital and ICU stay. Despite a higher intrinsic cost, linezolid has a better economic and clinical profile than vancomycin, both in ICU and non-ICU patients [20, 102,103,104,105,106,107]. Because pneumonia treatment duration dose not exceed 14 days, the side effects of linezolid are rarely observed during pneumonia treatment. In the 2016 American guidelines (Infectious Disease Society of America and American Thoracic Society) for the treatment of hospital-acquired or ventilator-associated pneumonia, the experts recommend to treat hospital-acquired or ventilator-associated pneumonia with vancomycin or linezolid rather than other antibiotics or antibiotic combinations (strong recommendation, moderate-quality evidence) [108]. The choice between vancomycin and linezolid should be guided by patient-specific factors such as blood cell counts, concurrent prescriptions for serotonin-reuptake inhibitors and renal function [108].
3.1.8.3 Acute Bacterial Skin and Skin Structure Infections
As for pneumonia, despite the theoretical advantages of oxazolidinones, the superiority of linezolid over vancomycin during the treatment of ABSSSIs is not clearly demonstrated in terms of outcomes. Numerous randomised trials and meta-analyses have been published on this issue.
One recent meta-analysis suggested equivalence of clinical efficacy between vancomycin, daptomycin and linezolid for the treatment of ABSSSIs [109]. Nevertheless, the authors reported a wide uncertainty margin, indicating the heterogeneity of the available evidence and the need for further research [109]. In contrast, several meta-analyses suggest a better efficacy and tolerance of linezolid compared with vancomycin for the treatment of ABSSSIs [110,111,112,113,114,115].
A recent comparison of the available meta-analyses (meta-analysis of the meta-analyses) suggests that linezolid could be associated with a shorter intravenous course and shorter hospital stay. However, because of the heterogeneity of available data and sponsoring by the pharmaceutical company producing linezolid, the data are not considered sufficient enough to support the widespread use of linezolid over vancomycin [111, 116]. The Infectious Disease Society of America guidelines suggest linezolid as an alternative to glycopeptides for the treatment of MRSA ABSSSIs, taking into account a patient’s condition, especially renal failure and blood cell count [117, 118]. Finally, despite an intrinsic higher cost, linezolid could be more cost effective than vancomycin or daptomycin for the treatment of ABSSSIs, especially because of a better tolerance [119, 120].
3.1.8.4 Skeletal Infections
Despite its excellent bone diffusion, there is a lack of evidence to support the routine use of linezolid for skeletal infections, mainly owing to the side effects of long-term linezolid treatment [121]. In the 2013 Infectious Disease Society of America guidelines for prosthetic joint infection management, vancomycin remains the first-line drug when MRSA is suspected [122]. Linezolid is the first alternative drug when vancomycin cannot be used [121, 122].
3.1.8.5 Central Nervous System Infections
Owing to the bacteriostatic activity of oxazolidinones, its class is not the first choice for CNS infection treatment. Nevertheless, because of its excellent distribution into several compartments, linezolid can be considered for CNS infection. In healthcare-associated meningitis and ventriculitis, vancomycin remains the first therapeutic step but linezolid is strongly recommended, despite low evidence, as long as vancomycin cannot be used [123]. In severe CNS infections, the standard dose of linezolid (600 mg twice daily) can be insufficient to achieve adequate CNS concentrations [31].
3.1.8.6 Bloodstream Infections
Owing to the bacteriostatic activity of oxazolidinones, its class is not recommended for BSI treatment. A meta-analysis has suggested that linezolid was non-inferior to vancomycin in staphylococci BSIs [124]. Nevertheless, one study has reported a higher mortality rate in catheter-related BSIs treated with linezolid when considering both Gram-negative and Gram-positive pathogens; this effect was no longer observed in the Gram-positive infection group [125]. However, linezolid could be effective to treat vancomycin-resistant enterococci BSI [126, 127]. Despite promising experimental data, oxazolidinones are not currently indicated for endocarditis related to vancomycin-susceptible strains, especially when daptomycin can be used [127, 128].
3.1.8.7 Special Populations
3.1.8.7.1 Critically Ill Patients and Renal Failure
In stable patients, no dose adjustment is necessary when renal dysfunction occurs. In patients receiving long-term dialysis, linezolid must be administered after dialysis session. In critically ill patients, commonly observed acute kidney injury (increasing the risk of drug accumulation) and increased volume of distribution (risk of subtherapeutic drug concentrations) both make the right dose for the right patient difficult to predict [129,130,131]. Variable linezolid exposure has been reported in this population [132]. In critically ill patients with renal replacement therapy, continuous modalities (continuous renal replacement therapy) are mainly used given their haemodynamic stability [133]. Moreover, continuous techniques provide a continuous clearance, resulting in more stable and predictable PK of drugs, especially antibiotics [133,134,135,136,137,138]. During continuous renal replacement therapy, there is a profound PK variability of linezolid exposing patients to underdosing [139]. Suboptimal achievement of therapeutic targets occurs at the EUCAST breakpoint MIC of 2 mg/L using intravenous 600 mg 12 hourly, especially for patients with a body weight > 90 kg [140]. In such patients, Monte Carlo simulations suggest a dosing regimen of 900 mg 12 hourly as the optimal dosing [140].
3.1.8.7.2 Obese Patients
It is classically admitted that drug dosing adjustments based on body mass index alone are not required in obese patients [16, 141, 142]. Standard doses for patients with body weights up to approximately 150 kg should provide AUC values similar to those seen in non-obese patients [141]. Nevertheless, despite their small sample size, some studies suggest that the optimal dosing regimen in obese patients could be 600 mg 6–8 hourly [143], in particular when less susceptible (MIC ≥ 2 or ≥ 4 mg/L) strains of S. aureus are suspected [144]. Monitoring of trough concentrations if clinical failure occurs is probably the only way to identify patients requiring higher doses.
4 Tedizolid: A Newer Oxazolidinone
Tedizolid phosphate (formerly TR-701 or torezolid phosphate) is a novel expanded-spectrum oxazolidinone with potent activity against a wide range of Gram-positive pathogens, including MRSA, VRE, S. pneumoniae, beta-hemolytic streptococci, viridans group streptococci and some linezolid-resistant strains [145, 146] (Table 3). Similarly to linezolid, tedizolid inhibits protein synthesis by binding the 23S ribosomal RNA of the 50S subunit. Tedizolid differs from other oxazolidinones by its modified side chain at the C5 position of the oxazolidinone nucleus, conferring its activity against some linezolid-resistant strains harbouring the resistant cfr gene [147]. Tedizolid exhibits a favourable pharmacokinetic profile with a biologically prolonged half-life, allowing once-daily administration and a more favourable haematological tolerability profile.
4.1 Tedizolid Pharmacokinetics
4.1.1 Pharmacokinetics Profile
Tedizolid phosphate is rapidly converted by apical alkaline phosphatases into the microbiologically active drug, tedizolid (Table 1). The addition of the phosphate group improves water solubility and provides excellent bioavailability. Indeed, the bioavailability of tedizolid after a single oral 200-mg dose was 91.5% [148]. Tedizolid also has minimal accumulation at steady state (~ 3 days) and has the benefit of a prolonged half-life allowing for once-daily dosing [149]. The pharmacokinetic profile in healthy volunteers is similar after single and multiple daily doses suggesting linear PK of tedizolid [149]. The mean AUC from time zero to the time of the last collected sample was 30 (± 10.3) mg·h/L, the mean maximum plasma concentration was 2.6 (± 0.6) mg/L and the mean half-life was 11 (± 0.8) h after a single intravenous 200-mg dose in healthy volunteers [148]. After analysing data from seven clinical trials, Flanagan et al. determined that the PK of tedizolid fits a two-compartment model with sigmoidal absorption, absolute bioavailability and linear elimination [150]. No clinical covariates affecting tedizolid PK were identified. Tedizolid is primarily metabolised by the liver, only 18% of the administered drug is eliminated in urine.
4.1.2 Tissue Penetration
4.1.2.1 Intrapulmonary Penetration
Pulmonary disposition of tedizolid following administration of 200 mg of oral tedizolid phosphate once daily was determined in healthy volunteers [151]. The calculated AUC0–24 values for ELF and alveolar macrophages were 109.3 and 52.95 mg·h/L, respectively. The ratio of penetration into the epithelial lining fluid and alveolar macrophages using the AUC0–24 in the respective compartment compared with ∫AUC0–24 in plasma were 40 and 20, respectively [151].
4.1.2.2 Skin and Soft-Tissue Penetration
In twelve healthy volunteers, tedizolid distribution into the ISF of subcutaneous adipose and skeletal muscle tissues was determined using microdialysis probes [152]. The mean ratios of free AUC in tissues over free AUC in plasma (∫AUCtissue/∫AUCplasma) were 1.1 ± 0.2 and 1.2 ± 0.2 for adipose and muscle tissue, respectively [152].
4.2 Tedizolid Pharmacodynamics
Based on MIC50 and MIC90 values, tedizolid is four- to eight-fold more potent than linezolid against all species of staphylococci, enterococci and streptococci, including drug-resistant phenotypes such as MRSA, VRSA and VRE [146]. A neutropenic mouse thigh model determined that the free-drug AUC over 24 h at steady state over the MIC (∫AUC0–24h/MIC) best correlated with efficacy against MRSA and methicillin-susceptible S. aureus [153]. The target total and free drug AUC/MIC ratios were calculated as 250 and approximately 50 (considering a protein binding of 80%), respectively [3, 153]. In non-neutropenic animals, the presence of granulocytes considerably increased the activity of tedizolid and lower exposures were required compared with neutropenic cohorts [154]. To achieve stasis in immunocompetent mice, the target ∫AUC/MIC was 3 or total AUC/MIC target of 15 (80% protein binding).
4.3 Tedizolid Dosing Considerations and Therapeutic Drug Monitoring
Unfortunately, to date, no information is available on tedizolid TDM and dosing optimisation.
4.4 Resistance to Tedizolid
As mentioned above, resistance to oxazolidinones is mainly the result of chromosomal mutations affecting the 23S ribosomal RNA or ribosomal proteins. Thus, pathogens resistant to linezolid owing to mutations in genes encoding 23S ribosomal RNA or ribosomal proteins are generally cross resistant to tedizolid. Resistance to linezolid can also be conferred by the plasmid-borne cfr gene. However, the hydroxymethyl group at the C5 position of the A-ring of tedizolid (which differs from the linezolide acetamide group at the C5 position) has been shown to maintain tedizolid potent activity against certain linezolid-resistant MRSA strains harbouring the cfr gene (in the absence of certain chromosomal mutations conferring reduced oxazolidinone susceptibility) [155,156,157].
4.5 Tedizolid Safety Profile and Adverse Events
4.5.1 Haematological Toxicity
From data of the two phase III, randomised double-blind clinical trials (ESTABLISH-1, ESTABLISH-2), tedizolid seems to confer a lower platelet reduction than linezolid (6.4% for the tedizolid phosphate group vs. 12.6% for the linezolid group) [158, 159]. In healthy volunteers treated with tedizolid for 21 days, no haematological side effects were observed [74]. However, additional data are required especially in at-risk populations treated with prolonged tedizolid therapy.
4.5.2 Neurological Toxicity
Preliminary data from a murine model do not provide any evidence for peripheral and optic neuropathy with tedizolid phosphate at plasma tedizolid exposures well above (approximately eightfold) the human therapeutic exposure levels [3]. The animal study involved 9 months of dosing as compared with 6 months in the study in which neuropathy findings were observed with linezolid [3]. Phase III trials including ABSSSI patients with a treatment period of 6 days revealed comparable incidences of peripheral or optic neuropathy between the tedizolid phosphate and the linezolid arms (peripheral neuropathy 1.2 vs. 0.6% for tedizolid phosphate and linezolid, respectively; optic nerve disorders 0.3 vs. 0.2%, respectively) [158, 159].
4.5.3 Lactic Acidosis
Recent experimental studies suggest that the mitochondrial toxicity of tedizolid could be less than linezolid, but this needs further investigations [71].
4.5.4 Gastrointestinal Toxicity
Fewer gastrointestinal side effects (16% for the tedizolid phosphate group vs. 23% for the linezolid group) have been reported in the two phase III, randomised clinical trials [160].
4.6 Tedizolid Drug Interactions
Tedizolid exhibits weak reversible inhibition of MAO in vitro [161]. Monoamine oxidase inhibitors co-administered with vasoconstrictors such as tyramine or pseudoephedrine can cause blood pressure elevations and induce hypertensive crises. Co-administration with serotoninergic agents may lead to potentially life-threatening serotonin syndrome. The potential interaction between tedizolid and oral tyramine and pseudoephedrine has been investigated in two randomised placebo-controlled trials [161]. Provocative testing in humans and animal models failed to demonstrate any potential for hypertensive or serotonergic adverse consequences at the therapeutic dose of tedizolid phosphate [161].
4.7 Tedizolid Clinical Indications
4.7.1 Acute Bacterial Skin and Skin Structure Infections
This new oxazolidinone has been approved on the basis of two phase III clinical trials (ESTABLISH-1, ESTABLISH-2) showing the non-inferiority of a 6-day course of 200 mg of tedizolid given once daily compared with a 10-day course of 600 mg linezolid given twice daily in ABSSSIs [158, 159].
4.7.2 Pneumonia
Currently, a phase III study is investigating the efficacy of 7 days of tedizolid in presumed Gram-positive healthcare-associated pneumonia or ventilator-associated pneumonia [Tedizolid Phosphate (TR-701 FA) vs Linezolid for the Treatment of Nosocomial Pneumonia (MK-1986-002), NCT02019420].
4.7.3 Special Populations
4.7.3.1 Renal and Hepatic Impairment
In renally and hepatically impaired patients, no dose adjustment seems necessary [162]. The mean AUC from time zero to the time of the last collected sample in the control group was 32.02 ( ± 9.32) mg·h/L compared with 29.69 ( ± 8.93) mg·h/L in the severe renal impairment group and 34.80 ( ± 20.72) mg·h/L in the hepatic impairment group. Haemodialysis does not require additional dosing considerations as, conversely to linezolid, tedizolid is highly protein bound ( ~ 80–90%).
4.7.3.2 Obese Patients
A retrospective analysis of phase I studies investigated the PK of tedizolid in obese (body mass index ≥ 30 kg/m2) patients and reported similar PK to those observed in non-obese patients, regardless of the degree of obesity. The authors conclude that no dose adjustment is required in such a population [163].
Several unaddressed questions regarding tedizolid PK remain, especially in populations such as critically ill patients and patients with hypoalbuminemia. Moreover, other oxazolidinones are currently under active investigation [164].
5 Conclusion
Antimicrobial resistance in Gram-positive pathogens is a major healthcare concern. In this context, oxazolidinones appear an effective alternative to vancomycin to treat MDR Gram-positive bacteria infections. Linezolid has been the only oxazolidinone approved for 14 years. Even though emergence of resistance to this antimicrobial agent remains relatively slow and stable over time, linezolid-resistant Gram-positive strains could become a future issue. Tedizolid, a newer oxazolidinone, may provide an effective treatment for linezolid-resistant strains and may overcome some of the limitations reported for linezolid. Its pharmacokinetic profile allows single daily dosing that can improve compliance in an outpatient setting and its safety profile appears better than linezolid in healthy volunteers with less myelosuppression. Further investigations are needed to determine the place of tedizolid in the antimicrobial arsenal.
References
Johnson AP, Warner M, Livermore DM. Activity of linezolid against multi-resistant gram-positive bacteria from diverse hospitals in the United Kingdom. J Antimicrob Chemother. 2000;45(2):225–30.
Full prescribing information: Sivextro®. 2015. http://sivextro.com/pdf/sivextro-prescribing-info.pdf. Accessed 7 May 2017.
European public assessment report summary for the public: Sivextro®. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/002846/WC500184804.pdf. Accessed 7 May 2017.
Mutnick AH, Biedenbach DJ, Turnidge JD, Jones RN. Spectrum and potency evaluation of a new oxazolidinone, linezolid: report from the SENTRY Antimicrobial Surveillance Program, 1998–2000. Diagn Microbiol Infect Dis. 2002;43(1):65–73.
Rybak MJ, Cappelletty DM, Moldovan T, Aeschlimann JR, Kaatz GW. Comparative in vitro activities and postantibiotic effects of the oxazolidinone compounds eperezolid (PNU-100592) and linezolid (PNU-100766) versus vancomycin against Staphylococcus aureus, coagulase-negative staphylococci, Enterococcus faecalis, and Enterococcus faecium. Antimicrob Agents Chemother. 1998;42(3):721–4.
Brown-Elliott BA, Ward SC, Crist CJ, Mann LB, Wilson RW, Wallace RJ Jr. In vitro activities of linezolid against multiple Nocardia species. Antimicrob Agents Chemother. 2001;45(4):1295–7.
Dresser LD, Rybak MJ. The pharmacologic and bacteriologic properties of oxazolidinones, a new class of synthetic antimicrobials. Pharmacotherapy. 1998;18(3):456–62.
Gee T, Ellis R, Marshall G, Andrews J, Ashby J, Wise R. Pharmacokinetics and tissue penetration of linezolid following multiple oral doses. Antimicrob Agents Chemother. 2001;45(6):1843–6.
Slatter JG, Stalker DJ, Feenstra KL, Welshman IR, Bruss JB, Sams JP, et al. Pharmacokinetics, metabolism, and excretion of linezolid following an oral dose of [(14)C]linezolid to healthy human subjects. Drug Metab Dispos. 2001;29(8):1136–45.
Stalker DJ, Jungbluth GL, Hopkins NK, Batts DH. Pharmacokinetics and tolerance of single- and multiple-dose oral or intravenous linezolid, an oxazolidinone antibiotic, in healthy volunteers. J Antimicrob Chemother. 2003;51(5):1239–46.
Wiskirchen DE, Shepard A, Kuti JL, Nicolau DP. Determination of tissue penetration and pharmacokinetics of linezolid in patients with diabetic foot infections using in vivo microdialysis. Antimicrob Agents Chemother. 2011;55(9):4170–5.
Diekema DJ, Jones RN. Oxazolidinone antibiotics. Lancet. 2001;358(9297):1975–82.
Full prescribing information: Zyvox®. http://labeling.pfizer.com/ShowLabeling.aspx?id=649. Accessed 25 Jun 2015.
Dehghanyar P, Burger C, Zeitlinger M, Islinger F, Kovar F, Muller M, et al. Penetration of linezolid into soft tissues of healthy volunteers after single and multiple doses. Antimicrob Agents Chemother. 2005;49(6):2367–71.
Welshman IR, Sisson TA, Jungbluth GL, Stalker DJ, Hopkins NK. Linezolid absolute bioavailability and the effect of food on oral bioavailability. Biopharm Drug Dispos. 2001;22(3):91–7.
Meagher AK, Forrest A, Rayner CR, Birmingham MC, Schentag JJ. Population pharmacokinetics of linezolid in patients treated in a compassionate-use program. Antimicrob Agents Chemother. 2003;47(2):548–53.
Mandell LA, Wunderink R. Methicillin-resistant Staphylococcus aureus and community-acquired pneumonia: an evolving relationship. Clin Infect Dis. 2012;54(8):1134–6.
Griffin AT, Peyrani P, Wiemken TL, Ramirez JA, Arnold FW. Empiric therapy directed against MRSA in patients admitted to the intensive care unit does not improve outcomes in community-acquired pneumonia. Infection. 2013;41(2):517–23.
Self WH, Wunderink RG, Williams DJ, Zhu Y, Anderson EJ, Balk RA, et al. Staphylococcus aureus community-acquired pneumonia: prevalence, clinical characteristics, and outcomes. Clin Infect Dis. 2016;63(3):300–9.
Takada H, Hifumi T, Nishimoto N, Kanemura T, Yoshioka H, Okada I, et al. Linezolid versus vancomycin for nosocomial pneumonia due to methicillin-resistant Staphylococcus aureus in the elderly: a retrospective cohort analysis. Effectiveness of linezolid in the elderly. Am J Emerg Med. 2017;35(2):245–8.
Boselli E, Breilh D, Rimmele T, Djabarouti S, Saux MC, Chassard D, et al. Pharmacokinetics and intrapulmonary diffusion of levofloxacin in critically ill patients with severe community-acquired pneumonia. Crit Care Med. 2005;33(1):104–9.
Boselli E, Breilh D, Caillault-Sergent A, Djabarouti S, Guillaume C, Xuereb F, et al. Alveolar diffusion and pharmacokinetics of linezolid administered in continuous infusion to critically ill patients with ventilator-associated pneumonia. J Antimicrob Chemother. 2012;67(5):1207–10.
Luna CM, Bruno DA, Garcia-Morato J, Mann KC, Risso Patron J, Sagardia J, et al. Effect of linezolid compared with glycopeptides in methicillin-resistant Staphylococcus aureus severe pneumonia in piglets. Chest. 2009;135(6):1564–71.
De Pascale G, Fortuna S, Tumbarello M, Cutuli SL, Vallecoccia M, Spanu T, et al. Linezolid plasma and intrapulmonary concentrations in critically ill obese patients with ventilator-associated pneumonia: intermittent vs continuous administration. Intensive Care Med. 2015;41(1):103–10.
Nau R, Sorgel F, Eiffert H. Penetration of drugs through the blood–cerebrospinal fluid/blood–brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23(4):858–83.
Cottagnoud P, Gerber CM, Acosta F, Cottagnoud M, Neftel K, Tauber MG. Linezolid against penicillin-sensitive and -resistant pneumococci in the rabbit meningitis model. J Antimicrob Chemother. 2000;46(6):981–5.
Cabellos C, Garrigos C, Taberner F, Force E, Pachon-Ibanez ME. Experimental study of the efficacy of linezolid alone and in combinations against experimental meningitis due to Staphylococcus aureus strains with decreased susceptibility to beta-lactams and glycopeptides. J Infect Chemother. 2014;20(9):563–8.
Villani P, Regazzi MB, Marubbi F, Viale P, Pagani L, Cristini F, et al. Cerebrospinal fluid linezolid concentrations in postneurosurgical central nervous system infections. Antimicrob Agents Chemother. 2002;46(3):936–7.
Beer R, Engelhardt KW, Pfausler B, Broessner G, Helbok R, Lackner P, et al. Pharmacokinetics of intravenous linezolid in cerebrospinal fluid and plasma in neurointensive care patients with staphylococcal ventriculitis associated with external ventricular drains. Antimicrob Agents Chemother. 2007;51(1):379–82.
Myrianthefs P, Markantonis SL, Vlachos K, Anagnostaki M, Boutzouka E, Panidis D, et al. Serum and cerebrospinal fluid concentrations of linezolid in neurosurgical patients. Antimicrob Agents Chemother. 2006;50(12):3971–6.
Luque S, Grau S, Alvarez-Lerma F, Ferrandez O, Campillo N, Horcajada JP, et al. Plasma and cerebrospinal fluid concentrations of linezolid in neurosurgical critically ill patients with proven or suspected central nervous system infections. Int J Antimicrob Agents. 2014;44(5):409–15.
Zeitlinger BS, Zeitlinger M, Leitner I, Muller M, Joukhadar C. Clinical scoring system for the prediction of target site penetration of antimicrobials in patients with sepsis. Clin Pharmacokinet. 2007;46(1):75–83.
Majcher-Peszynska J, Haase G, Sass M, Mundkowski R, Pietsch A, Klammt S, et al. Pharmacokinetics and penetration of linezolid into inflamed soft tissue in diabetic foot infections. Eur J Clin Pharmacol. 2008;64(11):1093–100.
Buerger C, Plock N, Dehghanyar P, Joukhadar C, Kloft C. Pharmacokinetics of unbound linezolid in plasma and tissue interstitium of critically ill patients after multiple dosing using microdialysis. Antimicrob Agents Chemother. 2006;50(7):2455–63.
Thallinger C, Buerger C, Plock N, Kljucar S, Wuenscher S, Sauermann R, et al. Effect of severity of sepsis on tissue concentrations of linezolid. J Antimicrob Chemother. 2008;61(1):173–6.
Lovering AM, Zhang J, Bannister GC, Lankester BJ, Brown JH, Narendra G, et al. Penetration of linezolid into bone, fat, muscle and haematoma of patients undergoing routine hip replacement. J Antimicrob Chemother. 2002;50(1):73–7.
Traunmuller F, Schintler MV, Spendel S, Popovic M, Mauric O, Scharnagl E, et al. Linezolid concentrations in infected soft tissue and bone following repetitive doses in diabetic patients with bacterial foot infections. Int J Antimicrob Agents. 2010;36(1):84–6.
Kutscha-Lissberg F, Hebler U, Muhr G, Koller M. Linezolid penetration into bone and joint tissues infected with methicillin-resistant staphylococci. Antimicrob Agents Chemother. 2003;47(12):3964–6.
Rana B, Butcher I, Grigoris P, Murnaghan C, Seaton RA, Tobin CM. Linezolid penetration into osteo-articular tissues. J Antimicrob Chemother. 2002;50(5):747–50.
Metallidis S, Nikolaidis J, Lazaraki G, Koumentaki E, Gogou V, Topsis D, et al. Penetration of linezolid into sternal bone of patients undergoing cardiopulmonary bypass surgery. Int J Antimicrob Agents. 2007;29(6):742–4.
Papadopoulos A, Plachouras D, Giannitsioti E, Poulakou G, Giamarellou H, Kanellakopoulou K. Efficacy and tolerability of linezolid in chronic osteomyelitis and prosthetic joint infections: a case–control study. J Chemother. 2009;21(2):165–9.
Rayner CR, Baddour LM, Birmingham MC, Norden C, Meagher AK, Schentag JJ. Linezolid in the treatment of osteomyelitis: results of compassionate use experience. Infection. 2004;32(1):8–14.
Senneville E, Legout L, Valette M, Yazdanpanah Y, Beltrand E, Caillaux M, et al. Effectiveness and tolerability of prolonged linezolid treatment for chronic osteomyelitis: a retrospective study. Clin Ther. 2006;28(8):1155–63.
Rao N, Ziran BH, Hall RA, Santa ER. Successful treatment of chronic bone and joint infections with oral linezolid. Clin Orthop Relat Res. 2004;427:67–71.
Falagas ME, Manta KG, Ntziora F, Vardakas KZ. Linezolid for the treatment of patients with endocarditis: a systematic review of the published evidence. J Antimicrob Chemother. 2006;58(2):273–80.
Chiang FY, Climo M. Efficacy of linezolid alone or in combination with vancomycin for treatment of experimental endocarditis due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47(9):3002–4.
Jacqueline C, Batard E, Perez L, Boutoille D, Hamel A, Caillon J, et al. In vivo efficacy of continuous infusion versus intermittent dosing of linezolid compared to vancomycin in a methicillin-resistant Staphylococcus aureus rabbit endocarditis model. Antimicrob Agents Chemother. 2002;46(12):3706–11.
Bernardo K, Pakulat N, Fleer S, Schnaith A, Utermohlen O, Krut O, et al. Subinhibitory concentrations of linezolid reduce Staphylococcus aureus virulence factor expression. Antimicrob Agents Chemother. 2004;48(2):546–55.
Diep BA, Afasizheva A, Le HN, Kajikawa O, Matute-Bello G, Tkaczyk C, et al. Effects of linezolid on suppressing in vivo production of staphylococcal toxins and improving survival outcomes in a rabbit model of methicillin-resistant Staphylococcus aureus necrotizing pneumonia. J Infect Dis. 2013;208(1):75–82.
Stevens DL, Herr D, Lampiris H, Hunt JL, Batts DH, Hafkin B. Linezolid versus vancomycin for the treatment of methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis. 2002;34(11):1481–90.
Rubinstein E, Cammarata S, Oliphant T, Wunderink R, Linezolid Nosocomial Pneumonia Study Group. Linezolid (PNU-100766) versus vancomycin in the treatment of hospitalized patients with nosocomial pneumonia: a randomized, double-blind, multicenter study. Clin Infect Dis. 2001;32(3):402–12.
Wunderink RG, Rello J, Cammarata SK, Croos-Dabrera RV, Kollef MH. Linezolid vs vancomycin: analysis of two double-blind studies of patients with methicillin-resistant Staphylococcus aureus nosocomial pneumonia. Chest. 2003;124(5):1789–97.
Wunderink RG, Niederman MS, Kollef MH, Shorr AF, Kunkel MJ, Baruch A, et al. Linezolid in methicillin-resistant Staphylococcus aureus nosocomial pneumonia: a randomized, controlled study. Clin Infect Dis. 2012;54(5):621–9.
Ferrer MD, Rodriguez JC, Alvarez L, Artacho A, Royo G, Mira A. Effect of antibiotics on biofilm inhibition and induction measured by real-time cell analysis. J Appl Microbiol. 2017;122(3):640–50.
Andes D, van Ogtrop ML, Peng J, Craig WA. In vivo pharmacodynamics of a new oxazolidinone (linezolid). Antimicrob Agents Chemother. 2002;46(11):3484–9.
Rayner CR, Forrest A, Meagher AK, Birmingham MC, Schentag JJ. Clinical pharmacodynamics of linezolid in seriously ill patients treated in a compassionate use programme. Clin Pharmacokinet. 2003;42(15):1411–23.
Morata L, Cuesta M, Rojas JF, Rodriguez S, Brunet M, Casals G, et al. Risk factors for a low linezolid trough plasma concentration in acute infections. Antimicrob Agents Chemother. 2013;57(4):1913–7.
Pea F, Furlanut M, Cojutti P, Cristini F, Zamparini E, Franceschi L, et al. Therapeutic drug monitoring of linezolid: a retrospective monocentric analysis. Antimicrob Agents Chemother. 2010;54(11):4605–10.
Adembri C, Fallani S, Cassetta MI, Arrigucci S, Ottaviano A, Pecile P, et al. Linezolid pharmacokinetic/pharmacodynamic profile in critically ill septic patients: intermittent versus continuous infusion. Int J Antimicrob Agents. 2008;31(2):122–9.
Lopez-Garcia B, Luque S, Roberts JA, Grau S. Pharmacokinetics and preliminary safety of high dose linezolid for the treatment of Gram-positive bacterial infections. J Infect. 2015;71(5):604–7.
Pea F, Cojutti PG, Baraldo M. A 10-year experience of therapeutic drug monitoring (TDM) of linezolid in a hospital-wide population of patients receiving conventional dosing: is there enough evidence for suggesting TDM in the majority of patients? Basic Clin Pharmacol Toxicol. 2017;121(4):303–8.
Pea F, Viale P, Cojutti P, Del Pin B, Zamparini E, Furlanut M. Therapeutic drug monitoring may improve safety outcomes of long-term treatment with linezolid in adult patients. J Antimicrob Chemother. 2012;67(8):2034–42.
Jones RN, Fritsche TR, Sader HS, Ross JE. Zyvox annual appraisal of potency and spectrum program results for 2006: an activity and spectrum analysis of linezolid using clinical isolates from 16 countries. Diagn Microbiol Infect Dis. 2007;59(2):199–209.
Boak LM, Rayner CR, Grayson ML, Paterson DL, Spelman D, Khumra S, et al. Clinical population pharmacokinetics and toxicodynamics of linezolid. Antimicrob Agents Chemother. 2014;58(4):2334–43.
Matsumoto K, Shigemi A, Takeshita A, Watanabe E, Yokoyama Y, Ikawa K, et al. Analysis of thrombocytopenic effects and population pharmacokinetics of linezolid: a dosage strategy according to the trough concentration target and renal function in adult patients. Int J Antimicrob Agents. 2014;44(3):242–7.
Rybak JM, Roberts K. Tedizolid phosphate: a next-generation oxazolidinone. Infect Dis Ther. 2015;4(1):1–14.
Long KS, Poehlsgaard J, Kehrenberg C, Schwarz S, Vester B. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob Agents Chemother. 2006;50(7):2500–5.
Prystowsky J, Siddiqui F, Chosay J, Shinabarger DL, Millichap J, Peterson LR, et al. Resistance to linezolid: characterization of mutations in rRNA and comparison of their occurrences in vancomycin-resistant enterococci. Antimicrob Agents Chemother. 2001;45(7):2154–6.
Vazquez JA, Arnold AC, Swanson RN, Biswas P, Bassetti M. Safety of long-term use of linezolid: results of an open-label study. Ther Clin Risk Manag. 2016;12:1347–54.
Bialvaei ZA, Rahbar M, Yousefi M, Asgharzadeh M, Samadi Kafil H. Linezolid: a promising option in the treatment of Gram-positives. J Antimicrob Chemother. 2017;72(2):354–64.
Flanagan S, McKee EE, Das D, Tulkens PM, Hosako H, Fiedler-Kelly J, et al. Nonclinical and pharmacokinetic assessments to evaluate the potential of tedizolid and linezolid to affect mitochondrial function. Antimicrob Agents Chemother. 2015;59(1):178–85.
Tsuji Y, Holford NHG, Kasai H, Ogami C, Heo YA, Higashi Y, et al. Population pharmacokinetics and pharmacodynamics of linezolid-induced thrombocytopenia in hospitalized patients. Br J Clin Pharmacol. 2017;83(8):1758–72.
Tsuji Y, Hiraki Y, Matsumoto K, Mizoguchi A, Kobayashi T, Sadoh S, et al. Thrombocytopenia and anemia caused by a persistent high linezolid concentration in patients with renal dysfunction. J Infect Chemother. 2011;17(1):70–5.
Lodise TP, Bidell MR, Flanagan SD, Zasowski EJ, Minassian SL, Prokocimer P. Characterization of the haematological profile of 21 days of tedizolid in healthy subjects. J Antimicrob Chemother. 2016;71(9):2553–8.
Soriano A, Ortega M, Garcia S, Penarroja G, Bove A, Marcos M, et al. Comparative study of the effects of pyridoxine, rifampin, and renal function on hematological adverse events induced by linezolid. Antimicrob Agents Chemother. 2007;51(7):2559–63.
Kuter DJ, Tillotson GS. Hematologic effects of antimicrobials: focus on the oxazolidinone linezolid. Pharmacotherapy. 2001;21(8):1010–3.
French G. Safety and tolerability of linezolid. J Antimicrob Chemother. 2003;51(Suppl. 2):ii45–53.
Soriano A, Gomez J, Gomez L, Azanza JR, Perez R, Romero F, et al. Efficacy and tolerability of prolonged linezolid therapy in the treatment of orthopedic implant infections. Eur J Clin Microbiol Infect Dis. 2007;26(5):353–6.
Corallo CE, Paull AE. Linezolid-induced neuropathy. Med J Aust. 2002;177(6):332.
Bressler AM, Zimmer SM, Gilmore JL, Somani J. Peripheral neuropathy associated with prolonged use of linezolid. Lancet Infect Dis. 2004;4(8):528–31.
Rho JP, Sia IG, Crum BA, Dekutoski MB, Trousdale RT. Linezolid-associated peripheral neuropathy. Mayo Clin Proc. 2004;79(7):927–30.
Kishor K, Dhasmana N, Kamble SS, Sahu RK. Linezolid induced adverse drug reactions: an update. Curr Drug Metab. 2015;16(7):553–9.
Narita M, Tsuji BT, Yu VL. Linezolid-associated peripheral and optic neuropathy, lactic acidosis, and serotonin syndrome. Pharmacotherapy. 2007;27(8):1189–97.
Mehta S, Das M, Laxmeshwar C, Jonckheere S, Thi SS, Isaakidis P. Linezolid-associated optic neuropathy in drug-resistant tuberculosis patients in Mumbai, India. PLoS One. 2016;11(9):e0162138.
Leach KL, Swaney SM, Colca JR, McDonald WG, Blinn JR, Thomasco LM, et al. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Mol Cell. 2007;26(3):393–402.
De Vriese AS, Coster RV, Smet J, Seneca S, Lovering A, Van Haute LL, et al. Linezolid-induced inhibition of mitochondrial protein synthesis. Clin Infect Dis. 2006;42(8):1111–7.
Palenzuela L, Hahn NM, Nelson RP Jr, Arno JN, Schobert C, Bethel R, et al. Does linezolid cause lactic acidosis by inhibiting mitochondrial protein synthesis? Clin Infect Dis. 2005;40(12):e113–6.
Protti A, Ronchi D, Bassi G, Fortunato F, Bordoni A, Rizzuti T, et al. Changes in whole-body oxygen consumption and skeletal muscle mitochondria during linezolid-induced lactic acidosis. Crit Care Med. 2016;44(7):e579–82.
Zuccarini NS, Yousuf T, Wozniczka D, Rauf AA. Lactic acidosis induced by linezolid mimics symptoms of an acute intracranial bleed: a case report and literature review. J Clin Med Res. 2016;8(10):753–6.
Im JH, Baek JH, Kwon HY, Lee JS. Incidence and risk factors of linezolid-induced lactic acidosis. Int J Infect Dis. 2015;31:47–52.
Apodaca AA, Rakita RM. Linezolid-induced lactic acidosis. N Engl J Med. 2003;348(1):86–7.
Johnson PC, Vaduganathan M, Phillips KM, O’Donnell WJ. A triad of linezolid toxicity: hypoglycemia, lactic acidosis, and acute pancreatitis. Proc (Bayl Univ Med Cent). 2015;28(4):466–8.
Hoyo I, Martinez-Pastor J, Garcia-Ramiro S, Climent C, Brunet M, Cuesta M, et al. Decreased serum linezolid concentrations in two patients receiving linezolid and rifampicin due to bone infections. Scand J Infect Dis. 2012;44(7):548–50.
Pea F, Cadeo B, Cojutti PG, Pecori D, Bassetti M. Linezolid underexposure in a hypothyroid patient on levothyroxine replacement therapy: a case report. Ther Drug Monit. 2014;36(5):687–9.
Sakai Y, Naito T, Arima C, Miura M, Qin L, Hidaka H, et al. Potential drug interaction between warfarin and linezolid. Intern Med. 2015;54(5):459–64.
Kalil AC, Klompas M, Haynatzki G, Rupp ME. Treatment of hospital-acquired pneumonia with linezolid or vancomycin: a systematic review and meta-analysis. BMJ Open. 2013;3(10):e003912.
Wang Y, Zou Y, Xie J, Wang T, Zheng X, He H, et al. Linezolid versus vancomycin for the treatment of suspected methicillin-resistant Staphylococcus aureus nosocomial pneumonia: a systematic review employing meta-analysis. Eur J Clin Pharmacol. 2015;71(1):107–15.
Kalil AC, Murthy MH, Hermsen ED, Neto FK, Sun J, Rupp ME. Linezolid versus vancomycin or teicoplanin for nosocomial pneumonia: a systematic review and meta-analysis. Crit Care Med. 2010;38(9):1802–8.
Jiang H, Tang RN, Wang J. Linezolid versus vancomycin or teicoplanin for nosocomial pneumonia: meta-analysis of randomised controlled trials. Eur J Clin Microbiol Infect Dis. 2013;32(9):1121–8.
Caffrey AR, Morrill HJ, Puzniak LA, Laplante KL. Comparative effectiveness of linezolid and vancomycin among a national veterans affairs cohort with methicillin-resistant Staphylococcus aureus pneumonia. Pharmacotherapy. 2014;34(5):473–80.
Sinha Ray A, Haikal A, Hammoud KA, Yu AS. Vancomycin and the risk of AKI: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2016;11(12):2132–40.
Patel DA, Michel A, Stephens J, Weber B, Petrik C, Charbonneau C. An economic model to compare linezolid and vancomycin for the treatment of confirmed methicillin-resistant Staphylococcus aureus nosocomial pneumonia in Germany. Infect Drug Resist. 2014;7:273–80.
Patel DA, Shorr AF, Chastre J, Niederman M, Simor A, Stephens JM, et al. Modeling the economic impact of linezolid versus vancomycin in confirmed nosocomial pneumonia caused by methicillin-resistant Staphylococcus aureus. Crit Care. 2014;18(4):R157.
Shorr AF, Puzniak LA, Biswas P, Niederman MS. Predictors of clinical success in the treatment of patients with methicillin-resistant Staphylococcus aureus (MRSA) nosocomial pneumonia (NP). PLoS One. 2015;10(7):e0131932.
Niederman MS, Chastre J, Solem CT, Wan Y, Gao X, Myers DE, et al. Health economic evaluation of patients treated for nosocomial pneumonia caused by methicillin-resistant Staphylococcus aureus: secondary analysis of a multicenter randomized clinical trial of vancomycin and linezolid. Clin Ther. 2014;36(9):1233 e1–1243 e1.
Lin PC, Wang BC, Kim R, Magyar A, Lai CC, Yang YW, et al. Estimating the cost-effectiveness of linezolid for the treatment of methicillin-resistant Staphylococcus aureus nosocomial pneumonia in Taiwan. J Microbiol Immunol Infect. 2016;49(1):46–51.
Rello J, Bin C. Cost of nosocomial pneumonia: the example of vancomycin versus linezolid-shorter stay or fewer complications? Int J Infect Dis. 2016;51:1–3.
Kalil AC, Metersky ML, Klompas M, Muscedere J, Sweeney DA, Palmer LB, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):e61–111.
Thom H, Thompson JC, Scott DA, Halfpenny N, Sulham K, Corey GR. Comparative efficacy of antibiotics for the treatment of acute bacterial skin and skin structure infections (ABSSSI): a systematic review and network meta-analysis. Curr Med Res Opin. 2015;31(8):1539–51.
Yue J, Dong BR, Yang M, Chen X, Wu T, Liu GJ. Linezolid versus vancomycin for skin and soft tissue infections. Cochrane Database Syst Rev. 2013;9(7):CD008056.
Yue J, Dong BR, Yang M, Chen X, Wu T, Liu GJ. Linezolid versus vancomycin for skin and soft tissue infections. Cochrane Database Syst Rev. 2016;9(1):CD008056.
Bounthavong M, Hsu DI. Efficacy and safety of linezolid in methicillin-resistant Staphylococcus aureus (MRSA) complicated skin and soft tissue infection (cSSTI): a meta-analysis. Curr Med Res Opin. 2010;26(2):407–21.
Beibei L, Yun C, Mengli C, Nan B, Xuhong Y, Rui W. Linezolid versus vancomycin for the treatment of gram-positive bacterial infections: meta-analysis of randomised controlled trials. Int J Antimicrob Agents. 2010;35(1):3–12.
Dodds TJ, Hawke CI. Linezolid versus vancomycin for MRSA skin and soft tissue infections (systematic review and meta-analysis). ANZ J Surg. 2009;79(9):629–35.
Nemeth J, Oesch G, Kuster SP. Bacteriostatic versus bactericidal antibiotics for patients with serious bacterial infections: systematic review and meta-analysis. J Antimicrob Chemother. 2015;70(2):382–95.
Tsoulas C, Nathwani D. Review of meta-analyses of vancomycin compared with new treatments for Gram-positive skin and soft-tissue infections: are we any clearer? Int J Antimicrob Agents. 2015;46(1):1–7.
Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18–55.
Stevens DL, Bisno AL, Chambers HF, Dellinger EP, Goldstein EJ, Gorbach SL, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):e10–52.
Bounthavong M, Hsu DI. Cost-effectiveness of linezolid in methicillin-resistant Staphylococcus aureus skin and skin structure infections. Expert Rev Pharmacoecon Outcomes Res. 2012;12(6):683–98.
Bounthavong M, Zargarzadeh A, Hsu DI, Vanness DJ. Cost-effectiveness analysis of linezolid, daptomycin, and vancomycin in methicillin-resistant Staphylococcus aureus: complicated skin and skin structure infection using Bayesian methods for evidence synthesis. Value Health. 2011;14(5):631–9.
Falagas ME, Siempos II, Papagelopoulos PJ, Vardakas KZ. Linezolid for the treatment of adults with bone and joint infections. Int J Antimicrob Agents. 2007;29(3):233–9.
Osmon DR, Berbari EF, Berendt AR, Lew D, Zimmerli W, Steckelberg JM, et al. Executive summary: diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):1–10.
Tunkel AR, Hasbun R, Bhimraj A, Byers K, Kaplan SL, Michael Scheld W, et al. Infectious Diseases Society of America’s Clinical Practice Guidelines for Healthcare-Associated Ventriculitis and Meningitis. Clin Infect Dis. 2017;64(6):e34–65.
Shorr AF, Kunkel MJ, Kollef M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studies. J Antimicrob Chemother. 2005;56(5):923–9.
Wilcox MH, Tack KJ, Bouza E, Herr DL, Ruf BR, Ijzerman MM, et al. Complicated skin and skin-structure infections and catheter-related bloodstream infections: noninferiority of linezolid in a phase 3 study. Clin Infect Dis. 2009;48(2):203–12.
Whang DW, Miller LG, Partain NM, McKinnell JA. Systematic review and meta-analysis of linezolid and daptomycin for treatment of vancomycin-resistant enterococcal bloodstream infections. Antimicrob Agents Chemother. 2013;57(10):5013–8.
Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52(3):285–92.
Baddour LM, Wilson WR, Bayer AS, Fowler VG Jr, Tleyjeh IM, Rybak MJ, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132(15):1435–86.
Roberts JA, Kumar A, Lipman J. Right dose, right now: customized drug dosing in the critically ill. Crit Care Med. 2017;45(2):331–6.
Roberts JA, Abdul-Aziz MH, Lipman J, Mouton JW, Vinks AA, Felton TW, et al. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis. 2014;14(6):498–509.
Tsai D, Lipman J, Roberts JA. Pharmacokinetic/pharmacodynamic considerations for the optimization of antimicrobial delivery in the critically ill. Curr Opin Crit Care. 2015;21(5):412–20.
Zoller M, Maier B, Hornuss C, Neugebauer C, Dobbeler G, Nagel D, et al. Variability of linezolid concentrations after standard dosing in critically ill patients: a prospective observational study. Crit Care. 2014;18(4):R148.
Neri M, Villa G, Garzotto F, Bagshaw S, Bellomo R, Cerda J, et al. Nomenclature for renal replacement therapy in acute kidney injury: basic principles. Crit Care. 2016;20(1):318.
KDIGO. Clinical practice guidelines for acute kidney injury. 2012. http://www.kdigo.org/clinical_practice_guidelines/AKI.php. Accessed 17 Sept 2017.
Jamal JA, Mueller BA, Choi GY, Lipman J, Roberts JA. How can we ensure effective antibiotic dosing in critically ill patients receiving different types of renal replacement therapy? Diagn Microbiol Infect Dis. 2015;82(1):92–103.
Roger C, Wallis SC, Muller L, Saissi G, Lipman J, Lefrant JY, et al. Influence of renal replacement modalities on Amikacin population pharmacokinetics in critically ill patients on continuous renal replacement therapy. Antimicrob Agents Chemother. 2016;60(8):4901–9.
Roger C, Wallis SC, Muller L, Saissi G, Lipman J, Bruggemann RJ, et al. Caspofungin population pharmacokinetics in critically ill patients undergoing continuous veno-venous haemofiltration or haemodiafiltration. Clin Pharmacokinet. 2017;56(9):1057–68.
Roger C, Wallis SC, Louart B, Lefrant JY, Lipman J, Muller L, et al. Comparison of equal doses of continuous venovenous haemofiltration and haemodiafiltration on ciprofloxacin population pharmacokinetics in critically ill patients. J Antimicrob Chemother. 2016;71(6):1643–50.
Villa G, Di Maggio P, De Gaudio AR, Novelli A, Antoniotti R, Fiaccadori E, et al. Effects of continuous renal replacement therapy on linezolid pharmacokinetic/pharmacodynamics: a systematic review. Crit Care. 2016;20(1):374.
Roger C, Muller L, Wallis SC, Louart B, Saissi G, Lipman J, et al. Population pharmacokinetics of linezolid in critically ill patients on renal replacement therapy: comparison of equal doses in continuous venovenous haemofiltration and continuous venovenous haemodiafiltration. J Antimicrob Chemother. 2016;71(2):464–70.
Bhalodi AA, Papasavas PK, Tishler DS, Nicolau DP, Kuti JL. Pharmacokinetics of intravenous linezolid in moderately to morbidly obese adults. Antimicrob Agents Chemother. 2013;57(3):1144–9.
Srinivas NR. Influence of morbidly obesity on the clinical pharmacokinetics of various anti-infective drugs: reappraisal using recent case studies-issues, dosing implications, and considerations. Am J Ther. 2016. doi:10.1097/MJT.0000000000000401.
Corcione S, Pagani N, Baietto L, Fanelli V, Urbino R, Ranieri VM, et al. Pharmacokinetics of high dosage of linezolid in two morbidly obese patients. J Antimicrob Chemother. 2015;70(10):2925.
Stein GE, Schooley SL, Peloquin CA, Kak V, Havlichek DH, Citron DM, et al. Pharmacokinetics and pharmacodynamics of linezolid in obese patients with cellulitis. Ann Pharmacother. 2005;39(3):427–32.
Pfaller MA, Flamm RK, Jones RN, Farrell DJ, Mendes RE. Activities of tedizolid and linezolid determined by the reference broth microdilution method against 3,032 Gram-positive bacterial isolates collected in Asia-Pacific, Eastern Europe, and Latin American countries in 2014. Antimicrob Agents Chemother. 2016;60(9):5393–9.
Chen KH, Huang YT, Liao CH, Sheng WH, Hsueh PR. In vitro activities of tedizolid and linezolid against Gram-positive cocci associated with acute bacterial skin and skin structure infections and pneumonia. Antimicrob Agents Chemother. 2015;59(10):6262–5.
Chen H, Yang Q, Zhang R, He W, Ma X, Zhang J, et al. In vitro antimicrobial activity of the novel oxazolidinone tedizolid and comparator agents against Staphylococcus aureus and linezolid-resistant Gram-positive pathogens: a multicentre study in China. Int J Antimicrob Agents. 2014;44(3):276–7.
Flanagan S, Fang E, Munoz KA, Minassian SL, Prokocimer PG. Single- and multiple-dose pharmacokinetics and absolute bioavailability of tedizolid. Pharmacotherapy. 2014;34(9):891–900.
Flanagan SD, Bien PA, Munoz KA, Minassian SL, Prokocimer PG. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy. 2014;34(3):240–50.
Flanagan S, Passarell J, Lu Q, Fiedler-Kelly J, Ludwig E, Prokocimer P. Tedizolid population pharmacokinetics, exposure response, and target attainment. Antimicrob Agents Chemother. 2014;58(11):6462–70.
Housman ST, Pope JS, Russomanno J, Salerno E, Shore E, Kuti JL, et al. Pulmonary disposition of tedizolid following administration of once-daily oral 200-milligram tedizolid phosphate in healthy adult volunteers. Antimicrob Agents Chemother. 2012;56(5):2627–34.
Sahre M, Sabarinath S, Grant M, Seubert C, Deanda C, Prokocimer P, et al. Skin and soft tissue concentrations of tedizolid (formerly torezolid), a novel oxazolidinone, following a single oral dose in healthy volunteers. Int J Antimicrob Agents. 2012;40(1):51–4.
Louie A, Liu W, Kulawy R, Drusano GL. In vivo pharmacodynamics of torezolid phosphate (TR-701), a new oxazolidinone antibiotic, against methicillin-susceptible and methicillin-resistant Staphylococcus aureus strains in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(7):3453–60.
Drusano GL, Liu W, Kulawy R, Louie A. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(11):5300–5.
Livermore DM, Mushtaq S, Warner M, Woodford N. Activity of oxazolidinone TR-700 against linezolid-susceptible and -resistant staphylococci and enterococci. J Antimicrob Chemother. 2009;63(4):713–5.
Locke JB, Morales G, Hilgers M, Kedar GC, Rahawi S, Jose Picazo J, et al. Elevated linezolid resistance in clinical cfr-positive Staphylococcus aureus isolates is associated with co-occurring mutations in ribosomal protein L3. Antimicrob Agents Chemother. 2010;54(12):5352–5.
Shaw KJ, Poppe S, Schaadt R, Brown-Driver V, Finn J, Pillar CM, et al. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob Agents Chemother. 2008;52(12):4442–7.
Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14(8):696–705.
Prokocimer P, De Anda C, Fang E, Mehra P, Das A. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA. 2013;309(6):559–69.
Shorr AF, Lodise TP, Corey GR, De Anda C, Fang E, Das AF, et al. Analysis of the phase 3 ESTABLISH trials of tedizolid versus linezolid in acute bacterial skin and skin structure infections. Antimicrob Agents Chemother. 2015;59(2):864–71.
Flanagan S, Bartizal K, Minassian SL, Fang E, Prokocimer P. In vitro, in vivo, and clinical studies of tedizolid to assess the potential for peripheral or central monoamine oxidase interactions. Antimicrob Agents Chemother. 2013;57(7):3060–6.
Flanagan S, Minassian SL, Morris D, Ponnuraj R, Marbury TC, Alcorn HW, et al. Pharmacokinetics of tedizolid in subjects with renal or hepatic impairment. Antimicrob Agents Chemother. 2014;58(11):6471–6.
Flanagan S, Minassian SL, Passarell JA, Fiedler-Kelly J, Prokocimer P. Pharmacokinetics of tedizolid in obese and nonobese subjects. J Clin Pharmacol. 2017;57(10):1290–4.
Phillips OA, Sharaf LH. Oxazolidinone antimicrobials: a patent review (2012–2015). Expert Opin Ther Pat. 2016;26(5):591–605.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this article. The authors acknowledge funding from the Australian National Health and Medical Research Council for a Centre of Research Excellence (APP1099452).
Conflict of interest
Jason A. Roberts is funded in part by a Practitioner Fellowship (APP1117065) from the National Health and Medical Research Council of Australia. Claire Roger and Laurent Muller have no conflicts of interest directly relevant to the content of this article.
Author Contributions
CR made substantial contributions to the literature search, interpretation and synthesis of published data, drafting the manuscript and approved the final version to be published. JAR made substantial contributions to revising the manuscript for important intellectual content and approved the final version to be published. LM made substantial contributions to the literature search, interpretation and synthesis of published data, drafting the manuscript and approved the final version to be published.
Rights and permissions
About this article

Cite this article
Roger, C., Roberts, J.A. & Muller, L. Clinical Pharmacokinetics and Pharmacodynamics of Oxazolidinones. Clin Pharmacokinet 57, 559–575 (2018). https://doi.org/10.1007/s40262-017-0601-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0601-x