
Abstract
Mutations in 12 different PEX genes can cause a generalized peroxisomal biogenesis disorder with clinical phenotypes ranging from Zellweger syndrome to infantile Refsum disease. To identify the specific PEX gene to be sequenced, complementation analysis is first performed in fibroblasts using catalase immunofluorescence. A patient with a relatively mild phenotype of infantile cholestasis, hypotonia and motor delay had elevated plasma very long-chain fatty acids and bile acid precursors, but fibroblast studies revealed normal or only mildly abnormal peroxisomal parameters and mosaic catalase immunofluorescence. This mosaicism persisted even when the incubation temperature was increased from 37 °C to 40 °C, a maneuver previously shown to abolish mosaicism by exacerbating peroxisomal dysfunction. As mosaicism precludes complementation analysis, a candidate gene approach was employed. After PEX1 sequencing was unrewarding, PEX12 sequencing revealed homozygosity for a novel c.102A>T (p.R34S) missense mutation affecting a partially conserved residue in the N-terminal region important for localization to peroxisomes. Transfection of patient fibroblasts with wild-type PEX12 cDNA confirmed that a PEX12 defect was the basis for the PBD. Homozygosity for c.102A>T was identified in a second patient of similar ethnic origin also presenting with a mild phenotype. PEX12 is a highly probable candidate gene for direct sequencing in the context of a mild clinical phenotype with mosaicism and minimally abnormal peroxisomal parameters in fibroblasts.
Similar content being viewed by others

Introduction
The generalized peroxisomal biogenesis disorders (PBDs) are a genetically heterogeneous group of rare, autosomal recessively inherited disorders (Gould et al. 2001). Their clinical manifestations include motor and cognitive neurological dysfunction, craniofacial dysmorphism, skeletal defects, liver disease, sensorineural deafness and retinopathy (Mandel and Korman 2003). The clinical severity of the generalized PBDs is quite variable and in fact represents a continuum—the Zellweger spectrum. Recognized phenotypes have conventionally been classified as Zellweger syndrome (OMIM #214100) at the most severe end of the spectrum through the intermediate neonatal adrenoleukodystrophy (NALD) (OMIM #202370) to the least severe infantile Refsum disease (IRD) (OMIM #266510).
The PBDs are caused by mutations in any of 12 genes from the PEX family of genes encoding the peroxins, a series of specialized proteins responsible for normal peroxisomal biogenesis. This is a highly complex process whereby peroxisomal membranes are assembled and then peroxisomal matrix proteins carrying a peroxisomal targeting sequence (PTS) at either their carboxy (PTS1) or amino (PTS2) terminus are targeted from the cytosol and imported into the organelle (Wanders and Waterham 2005).
The generalized PBDs are characterized by an absence of morphologically identifiable peroxisomes (or a presence of ghost-like residual structures) and a loss of multiple peroxisomal functions (both PTS1 and PTS2 matrix enzymes). The peroxisomal matrix normally contains more than 50 enzymes, which participate in a multitude of essential biosynthetic and catabolic functions. These include β-oxidation of very long-chain fatty acids (VLCFA) and bile acid precursors, α- and β-oxidation of the branched long-chain fatty acids (BCFA) phytanic acid and pristanic acid, respectively, formation of etherphospholipids (including plasmalogens), polyunsaturated fatty acids (PUFA), cholesterol, leukotrienes and glyoxylate, and metabolism of H2O2, glutaryl-CoA and pipecolic acid (Wanders 2004).
In a generalized PBD, it is not possible to predict which of the 12 candidate PEX genes is mutated on the basis of clinical or biochemical findings. Identification of the specific PEX gene involved is therefore achieved by performing complementation analysis. This entails generating hybrid cells by fusing the patient’s fibroblasts with a reference fibroblast line, for which the PEX defect has been defined. Restoration of peroxisomal function, i.e., complementation, manifests as the replacement of a diffuse pattern of catalase immunofluorescence staining (i.e., cytoplasmic location) by a punctate (i.e., peroxisomal location) pattern. Complementation implies that the defects in the patient and reference lines reside in different genes, whereas failure of complementation reveals the patient’s defect to be in the same PEX gene as the reference line (Yajima et al. 1992). Using this approach, the generalized PBDs have been divided into 12 distinct complementation groups (CG), and the underlying PEX gene has been identified for each of these groups.
We report a PBD patient with a relatively mild clinical phenotype and minimal abnormalities of peroxisomal function in fibroblasts and describe the identification of the underlying PEX gene mutation despite an inability to perform complementation analysis because of a mosaic pattern of catalase immunofluorescence. Subsequently, we identified the same mutation as the cause of a PBD in a second patient of similar ethnic origin who also presented with a relatively mild phenotype.
Materials and methods
Informed consent to perform the following studies was obtained.
Metabolite determinations
Urine organic acids were extracted from random urine specimens by a standard ethyl acetate/diethyl ether procedure, converted to their trimethylsilyl (TMS) derivatives, and analyzed on a Varian/Finnigan™ (Palo Alto, CA, USA) gas chromatography–mass spectrometry (GC–MS) system. VLCFA and BCFA levels in plasma were determined by GC/MS as their methyl derivatives with deuterated internal standards, essentially according to an established method (Vallance and Applegarth 1994).
Peroxisomal studies in fibroblasts
The following studies in cultured fibroblasts were performed according to published methods: VLCFA concentrations (Valianpour et al. 2003), C26:0 and pristanic acid β-oxidation (Wanders et al. 1995b), phytanic acid α-oxidation (Wanders and van Roermund 1993), immunoblot analysis of acyl-CoA oxidase and peroxisomal thiolase (Wanders et al. 1995a), dihydroxyacetonephosphate acyltransferase (DHAP-AT) activity and catalase immunofluorescence microscopy (Wanders et al. 1989).
To determine the abundance of peroxisomes at different temperatures, fibroblasts of the patient were cultured for two weeks at 30, 37 or 40 °C. Cells were subsequently transfected with the pEGFP-C3 vector (BD Biosciences, San Jose, CA, USA), in which we introduced the peroxisomal targeting sequence SKL carboxy-terminal of the encoded enhanced green fluorescent protein (EGFP-SKL), or with the pEGFP-SKL vector plus wild-type human PEX12 cDNA in pcDNA3 (Invitrogen, Carlsbad, CA, USA). The subcellular localization of EGFP-SKL was determined with fluorescence microscopy at 488 nm.
Mutation analysis
Mutation analysis was performed as previously described for PEX1 (Walter et al. 2001) and PEX12 (Gootjes et al. 2004b). PEX12 nucleotides are numbered from the first ATG codon according to the mRNA sequence of GenBank, accession number NM_000286.
Results
Patient 1 case description
The patient is the first-born son of healthy, apparently unrelated parents of Iranian Jewish origin. He was born at 39 weeks gestation by cesarean section because of breech presentation. His birth weight was 2.70 kg and Apgar scores were nine and ten at 1 and 5 min, respectively. During the first ten days of life he underwent phototherapy twice because of indirect hyperbilirubinemia that reached a maximum level of 357 μmol/L. He was hospitalized at age six weeks because of reappearance of jaundice and development of acholic stools. He was below the third percentile for weight, height and head circumference and had moderate hepatosplenomegaly as well as generalized hypotonia and severe head lag.
Plasma bilirubin level was 153 μmol/l with a conjugated fraction of 42 μmol/l. Liver enzymes were elevated, with AST 142 U/l (7–46), ALT 69 U/l (7–46), GGT 153 U/l (0–45) and alkaline phosphatase 575 U/l (100–300). He underwent a comprehensive evaluation for cholestatic jaundice of infancy. Abdominal ultrasonography showed normal bile ducts and a 99mTc-DIPA scan revealed delayed passage of the isotope into the gastrointestinal tract, consistent with parenchymal liver disease and excluding the possibility of extrahepatic biliary atresia. Serology for infectious causes (hepatitis A, B and C viruses, cytomegalovirus, Epstein–Barr virus, herpes simplex, rubella, toxoplasmosis, and syphilis) was negative. Thyroid function, α1-antitrypsin and galactosemia screen were all normal.
A urine organic acid analysis was requested to exclude hepatorenal tyrosinemia. Succinylacetone was not detected, but there was a prominent excretion of dicarboxylic acids including 3,6-epoxy derivates, in addition to 4-hydroxyphenyllactic and 2-hydroxyisovaleric acids. This profile was suggestive of a peroxisomal disorder (Korman et al. 2000) and therefore prompted analysis of VLCFA in plasma, followed by a complete profile of peroxisomal biochemical functions (below and Table 1), which confirmed the diagnosis of a PBD.
The hyperbilirubinemia resolved completely by age five months. Hypotonia persisted and his gross motor development was slow but progressive. He was able to roll over in both directions at age nine months, crawl at 15 months and walk with support at two years. Fine motor, social and language development were better preserved and at age two years he had good social skills and comprehension and spoke with single words. He had no evidence of a hearing deficit (normal auditory brainstem evoked response) or retinopathy (normal fundoscopic examination, electroretinogram and visual evoked potential).
Plasma and bloodspot metabolites
Plasma C26:0 levels and C26:0/C22:0 ratio and bloodspot bile acid intermediates (DHCA and THCA) were markedly elevated (Table 1), consistent with a defect in peroxisomal β-oxidation. The BCFA (phytanic and pristanic acids) were within normal limits (although even in ZS, elevation of BCFA may develop only after weeks or months, as phytanic acid is derived from exogenous dietary sources).
Peroxisomal studies in fibroblasts
Studies in fibroblasts (Table 1) revealed abnormalities in multiple peroxisomal functions, consistent with a generalized PBD. However, in contrast to the clear-cut metabolite abnormalities in urine organic acid and plasma VLCFA and bile acid testing, most of the fibroblast studies showed only borderline dysfunction or even normal results. The C26:0 level in fibroblasts was within normal limits, with a mildly elevated C26:0/C22:0 ratio. There was a partially deficient rate of C26:0 and pristanic acid β-oxidation and phytanic acid α-oxidation. DHAP-AT activity in fibroblasts (and also in platelets) was borderline low, whilst the DHAP-AT/GDH ratio was within normal limits. Immunoblotting for acyl-CoA yielded the normal 70, 50, and 20 kDa bands, whereas only the 70 kDa band would be expected in a typical PBD. Similarly, the normal 41 kDa and not the abnormal 44 kDa precursor form of peroxisomal thiolase was found.
Immunofluorescence microscopy using antibodies raised against the peroxisomal matrix enzyme, catalase, showed a mosaic pattern with some cells positive for peroxisomes (punctate staining) and others lacking peroxisomes (diffuse staining). However, in contrast to previous experience with a group of patients that had similar findings in fibroblasts (Gootjes et al. 2004c), this mosaic pattern persisted even when the cells were incubated at 40 °C. In these circumstances it was not possible to perform complementation analysis.
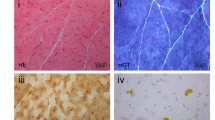
The mosaic pattern was also observed when the patient cells were transfected with a peroxisomal green fluorescent protein-SKL (EGFP-SKL) reporter. Using this reporter we observed that at 37 °C approximately 80% of the transfected cells still contained peroxisomes (Fig. 1b). When the cells were first cultured for two weeks at 40 °C and then transfected with the EGFP-SKL reporter, we still observed that ∼20% of the transfected cells contained peroxisomes, although the number of peroxisomes per cell was markedly lower (Fig. 1c). In addition, the cells looked rather bad under this condition. Surprisingly, when we cultured the cells for two weeks at 30 °C we only observed peroxisomes in ∼30% of the transfected cells (Fig. 1a). This contrasts with previous studies where we observed higher proportions of cells with peroxisomes when cultured at lower temperatures (Gootjes et al. 2004c).

Peroxisome abundance in fibroblasts of the patient. Fibroblasts of the patient were cultured for two weeks at 30 °C (a), 37 °C (b), and 40 °C (c, d) and subsequently transfected with the expression plasmid encoding EGFP-SKL. Cells in d were cotransfected with pcDNA3-PEX12. The subcellular localization (diffuse cytosolic or punctate peroxisomal) was visualized with fluorescence microscopy
PEX gene mutation analysis
Because the gene responsible for the PBD in this patient could not be identified by cell fusion complementation analysis, we elected to sequence candidate PEX genes (see “Discussion”). Sequencing of PEX1 did not reveal any mutation. We then proceeded to PEX12. Sequence analysis of PEX12 gDNA revealed the patient to be homozygous and his parents to be heterozygous for a novel c.102A>T nucleotide substitution. This changes codon 34 from AGA encoding arginine to AGT encoding serine (p.R34S).
To confirm that the peroxisomal abnormalities in the patient were indeed due to a defective PEX12 gene, we cotransfected cells of the patient, cultured for two weeks at 40 °C, with EGFP-SKL and wild-type PEX12 cDNA in pcDNA3 or empty pcDNA3. This resulted in punctate peroxisomal fluorescence in ∼ 60% of the cells transfected with EGFP-SKL plus PEX12 cDNA (Fig. 1d) when compared to ∼ 20% punctate fluorescence in the same cells transfected with EGFP-SKL and the empty pcDNA3 vector.
Patient 2
This patient is the first-born son of healthy, related parents of Iranian Jewish origin. The pregnancy was a product of in vitro fertilization. He was born at term weighing 2.90 kg with Apgar scores of seven and nine at 1 and 5 min, respectively. Hypotonia was noted in the post-natal period. Neonatal jaundice resolved spontaneously. He rolled over at six months and walked at 25 months. He was evaluated at age two years and ten months for hypotonia and delayed gross motor development. His cognitive development was completely normal for age. There was no facial or other dysmorphism. His gait was wide-based and he had a positive Gower’s sign. Deep tendon reflexes were absent in the lower limbs. Routine investigations revealed mildly elevated liver transaminases (AST 129 U/l, ALT 86 U/l) with normal total bilirubin concentration (7 μmol/l).
Urine organic acid profile was suggestive of a peroxisomal disorder with a marked excretion of 3,6-epoxy-dicarboxylic acids and increased C10 dicarboxylic (sebacic), 4-hydroxyphenyllactic and 2-hydroxyisovaleric acids. Determination of plasma VLCFA and BCFA revealed a marked elevation of plasma C26:0 levels and the C26:0/C22:0 ratio and a clear elevation of phytanic and pristanic acids (Table 1). Skin biopsy for fibroblast culture and further evaluation of peroxisomal functions was not performed. However, the common ethnic origin of Patients 1 and 2 (both Iranian Jewish) and their relatively mild clinical phenotypes raised the possibility that they shared an identical molecular basis for their peroxisomal defect. Indeed, PEX12 sequencing revealed Patient 2 to be homozygous and his parents heterozygous for the same c.102A>T mutation previously detected in Patient 1.
Discussion
PBDs are usually associated with major morbidity and mortality. The patients in the current report, however, have a somewhat milder phenotype than classical IRD. A number of such patients with relatively mild presentations or atypical features have been reported (Raas-Rothschild et al. 2002). In general, PBD patients with milder clinical manifestations tend to have less severe peroxisomal dysfunction on biochemical testing (Moser et al. 1995) and there is a correlation between life expectancy and the severity of impairment of DHAP-AT activity and C26:0 β-oxidation in fibroblasts (Gootjes et al. 2002). Two additional phenomena have been observed in cases with milder clinical and biochemical phenotypes. The first is mosaicism, the presence of peroxisomes in some cells but the absence of them in other cells of the same tissue, as detected by immunocytochemical or electron microscopy studies (Mandel et al. 1994; Roels et al. 2003). The second is temperature sensitivity, the restoration of morphological peroxisome formation and peroxisomal biochemical function in PBD fibroblasts cultured at 30 °C rather than 37 °C (Hashimoto et al. 2005; Imamura et al. 1998; Osumi et al. 2000). It has been suggested that temperature sensitivity is a consequence of an imbalance of folding and unfolding kinetics of mutant proteins, resulting in a reduction of correctly folded protein as temperature increases (Hashimoto et al. 2005).
The traditional approach to identifying the molecular defect in a PBD patient has been to first identify which one of the many PEX genes is involved by performing complementation analysis in fibroblasts and then to sequence that specific gene. However, the mosaic pattern of catalase immunofluorescence observed in the fibroblasts of Patient 1 precluded complementation analysis. This is because the marker which is used to indicate successful complementation is the appearance of punctate staining in fused hybrid cells, but this was already present in some of the patient’s cells even prior to complementation (mosaicism). Gootjes et al. (2004a) reasoned that, just as lowering the incubation temperature leads to improved peroxisomal function in temperature-sensitive PBD fibroblasts, increasing the temperature might exacerbate the defect and prevent peroxisome formation. Indeed, in a group of patients with fibroblast mosaicism, they found that raising the incubation temperature to 40 °C completely abolished the punctate pattern of catalase immunofluorescence that had been present in some cells. This maneuver thus overcomes the obstacle to complementation analysis, enabling the molecular basis to be determined (Gootjes et al. 2004a).
Nevertheless, in the current case, the mosaic pattern of catalase immunofluorescence persisted even at 40 °C. With no possibility of performing complementation analysis, we therefore elected to employ a candidate gene approach in order to identify the mutated PEX gene. We initially chose to screen for mutations in PEX1, the gene which is mutated in CG1. This is because CG1 accounts for more than half of all PBD patients and also because PEX1 mutations, particularly the G843D allele, have often been identified in PBD patients with prolonged survival and/or milder clinical phenotypes (Gartner et al. 1999; Poll-The et al. 2004; Reuber et al. 1997). When the PEX1 analysis was unrewarding, however, we next selected the PEX12 gene; mutations in this have been associated with milder clinical phenotypes and biochemical abnormalities in fibroblasts and also with mosaic catalase immunofluorescence (Gootjes et al. 2004a, 2004c). This led to the successful identification of the homozygous PEX12 c.102A>T (p.R34S) mutation in Patient 1, and subsequently homozygosity for c.102A>T in Patient 2. Transfection of Patient 1 fibroblasts with wild-type PEX12 cDNA confirmed that the peroxisomal abnormalities were indeed due to a PEX12 defect.
Patient 1 resembles the group of patients with a PEX12 defect described by Gootjes et al. (2004a), except that his clinical phenotype was even milder and the fibroblast mosaicism was not abolished at 40 °C. In this regard, he resembles more closely the patient originally thought to have trihydroxycholestanoyl-CoA oxidase deficiency and recently shown to have a PEX12 defect (Gootjes et al. 2004c). Taken together, these observations suggest that the combination of relatively mild clinical phenotype, minimal abnormalities in fibroblast peroxisomal biochemical functions, and a mosaic pattern of fibroblast catalase immunofluorescence is highly suggestive of a PEX12 defect. Our experience would suggest that PEX12 (all three exons) should be a prime candidate for mutation testing in such patients. Proceeding directly to PEX12 mutation analysis would be particularly efficacious in such patients for whom complementation analysis is difficult or impossible due to peroxisomal mosaicism.
An alternative approach to ascertaining the genetic defect in PBD patients without the need for preliminary complementation analysis has recently been proposed by Steinberg et al. (2004). They developed the “PEX Gene Screen,” a hierarchical algorithm consisting of seven sequential steps by which 12 individual exons from six different PEX genes are sequenced, these specific exons having been selected as the most frequently mutated according to published reports. According to their strategy, exons 2 and 3 of PEX12 are screened at the fifth step of the seven-step algorithm. Because the c.102A>T (p.R34S) mutation identified in our patient is located in exon 1 of PEX12, it would not have been detected by their strategy.
The PEX12 gene is located on chromosome 17q12 and comprises three exons, which encode peroxisomal biogenesis factor 12 (Pex12p), a 40.8 kDa protein consisting of 359 amino acids. It is an integral peroxisomal membrane protein with two transmembrane segments and has its N- and C-terminal parts exposed to the cytoplasm. The N-terminal region of Pex12p is required for its localization to peroxisomes (Okumoto et al. 2000). The C-terminal region contains an atypical RING finger zinc-binding domain of the C3HC4-type, which is important for its interaction with PEX5, the import receptor for PTS1-containing proteins, as well as PEX10 (Chang et al. 1999; Okumoto et al. 2000) and PEX19 (Sacksteder et al. 2000).
The c.102A>T (p.R34S) mutation associated with mild clinical phenotype and fibroblast abnormalities in the current report is located in the N-terminal region of the protein. This region is important for the localization of Pex12p to the peroxisomes. In a study of the intracellular localization and complementing activity of PEX12 N-terminal deletion mutants, a mutant with an internal deletion of codons 26–48 (i.e., including the arginine residue at codon 34) was not expressed and was not biologically active (Okumoto et al. 2000). Although this arginine residue at codon 34 is conserved in mammalian species as well as roundworm (C. elegans) and thale cress (Arabidopsis thaliana), it is not conserved in other organisms including yeasts (P. pastoris, P. angusta, S. cerevisiae, C. albicans), fruit fly (D. melanogaster) and zebra fish (B. rerio). The arginine at residue 34 may therefore not be critical for Pex12p function, and this could explain the relatively mild clinical features of the patients and the mild biochemical phenotype in fibroblasts.
A clear phenotype–genotype correlation has been demonstrated for PBD patients belonging to the CG3 group caused by PEX12 mutations (Chang and Gould 1998; Gootjes et al. 2004b). All patients with the severe ZS phenotype and survival <12 months had mutations resulting in truncation of the protein such that it lacks the COOH-terminal zinc-binding domain essential for its interaction with PEX5 and PEX10. In contrast, most patients with the milder phenotypes had mutations that produce a full-length protein. A patient with the NALD clinical phenotype and survival >4 years was homozygous for a 3-bp deletion c.202–204delCTT that removed the leucine at codon 68; this mutation does not completely eliminate Pex12p function (Chang and Gould 1998). A patient with the IRD clinical phenotype had a missense mutation c.273A>T (p.R91S) in the N-terminal part of the protein. Another patient with the IRD clinical phenotype and a survival of >23 years had an unusual mutation, which was predicted to truncate the protein only eight amino acids after initiation but which in fact produces a functional protein following reinitiation of translation at a downstream AUG codon (Chang and Gould 1998). Unlike the preceding mutations associated with a milder phenotype and located in the N-terminal part of the protein, the c.959C>T (p.S320F) found in the atypical patients with very mildly abnormal fibroblast functions is located in the C-terminal zinc-binding domain. However, the serine at position 320 is not present in yeast orthologs and therefore may not be critical to Pex12p function (Gootjes et al. 2004b). Another missense mutation, c.949C>T (p.L317F), found on one allele of another patient with minimal biochemical abnormalities, was also located in the same region (Gootjes et al. 2004c).
The observations in the present report have some important implications from the clinical and laboratory perspectives. A PBD should be always be included in the differential diagnosis of cholestatic liver disease during infancy, even if other typical features of an IRD phenotype such as gastrointestinal dysfunction, craniofacial dysmorphism, retinitis, and auditory impairment are lacking. Examination of plasma VLCFA should be incorporated into the protocol for investigating neonatal conjugated hyperbilirubinemia, together with the usual tests for anatomical, infectious, genetic, and other metabolic causes. In this context, it is important to be aware of the highly suggestive profile that is found upon the GC–MS examination of urine organic acids (Korman et al. 2000), an investigation which is often requested in this situation for the purpose of excluding hepatorenal tyrosinemia or other metabolic liver disease.
Another important issue in this and similar cases concerns the difficulty involved in providing prenatal diagnosis in future pregnancies. In general, prenatal diagnosis of PBD may be achieved by assaying peroxisomal biochemical functions (Schutgens et al. 1989; Steinberg et al. 2005; Wanders et al. 1991), or alternatively by mutation analysis—provided that the complementation group and specific gene defect have already been identified in the family (Wanders and Waterham 2005). Considering that fibroblasts from Patient 1 in this report exhibited normal or only marginally abnormal results for VLCFA, β-oxidation, DHAP-AT activity, and other peroxisomal functions, unequivocal prenatal diagnosis would not have been possible by biochemical analysis of CVS tissue. Furthermore, the complementation group and PEX gene responsible could not be identified because of the mosaic pattern of catalase immunofluorescence in fibroblasts, even with the modification where the cells were cultured at 40 °C (Gootjes et al. 2004a). The realization that PEX12 is a highly probable candidate gene for direct sequencing in the context of a relatively mild clinical phenotype with fibroblast mosaicism and only minimal abnormalities of peroxisomal parameters in fibroblasts can enable resolution of the molecular basis in such cases and permit prenatal diagnosis in future pregnancies.
References
Chang CC, Gould SJ (1998) Phenotype-genotype relationships in complementation group 3 of the peroxisome-biogenesis disorders. Am J Hum Genet 63:1294–1306
Chang CC, Warren DS, Sacksteder KA, Gould SJ (1999) PEX12 interacts with PEX5 and PEX10 and acts downstream of receptor docking in peroxisomal matrix protein import. J Cell Biol 147:761–774
Gartner J, Preuss N, Brosius U, Biermanns M (1999) Mutations in PEX1 in peroxisome biogenesis disorders: G843D and a mild clinical phenotype. J Inherit Metab Dis 22:311–313
Gootjes J, Mooijer PA, Dekker C, Barth PG, Poll-The BT, Waterham HR, Wanders RJ (2002) Biochemical markers predicting survival in peroxisome biogenesis disorders. Neurology 59:1746–1749
Gootjes J, Schmohl F, Mooijer PA, Dekker C, Mandel H, Topcu M, Huemer M, Von Schutz M, Marquardt T, Smeitink JA, Waterham HR, Wanders RJ (2004a) Identification of the molecular defect in patients with peroxisomal mosaicism using a novel method involving culturing of cells at 40°C: implications for other inborn errors of metabolism. Hum Mutat 24:130–139
Gootjes J, Schmohl F, Waterham HR, Wanders RJ (2004b) Novel mutations in the PEX12 gene of patients with a peroxisome biogenesis disorder. Eur J Hum Genet 12:115–120
Gootjes J, Skovby F, Christensen E, Wanders RJ, Ferdinandusse S (2004c) Reinvestigation of trihydroxycholestanoic acidemia reveals a peroxisome biogenesis disorder. Neurology 62:2077–2081
Gould SJ, Raymond GV, Valle D (2001) The peroxisome biogenesis disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th edn. McGraw–Hill, New York, pp 3181–3217
Hashimoto K, Kato Z, Nagase T, Shimozawa N, Kuwata K, Omoya K, Li A, Matsukuma E, Yamamoto Y, Ohnishi H, Tochio H, Shirakawa M, Suzuki Y, Wanders RJ, Kondo N (2005) Molecular mechanism of a temperature-sensitive phenotype in peroxisomal biogenesis disorder. Pediatr Res 58:263–269
Imamura A, Tamura S, Shimozawa N, Suzuki Y, Zhang Z, Tsukamoto T, Orii T, Kondo N, Osumi T, Fujiki Y (1998) Temperature-sensitive mutation in PEX1 moderates the phenotypes of peroxisome deficiency disorders. Hum Mol Genet 7:2089–2094
Korman SH, Mandel H, Gutman A (2000) Characteristic urine organic acid profile in peroxisomal biogenesis disorders. J Inherit Metab Dis 23:425–428
Mandel H, Espeel M, Roels F, Sofer N, Luder A, Iancu TC, Aizin A, Berant M, Wanders RJ, Schutgens RB (1994) A new type of peroxisomal disorder with variable expression in liver and fibroblasts. J Pediatr 125:549–555
Mandel H, Korman SH (2003) Phenotypic variability (heterogeneity) of peroxisomal disorders. Adv Exp Med Biol 544:9–30
Moser AB, Rasmussen M, Naidu S, Watkins PA, McGuinness M, Hajra AK, Chen G, Raymond G, Liu A, Gordon D (1995) Phenotype of patients with peroxisomal disorders subdivided into sixteen complementation groups. J Pediatr 127:13–22
Okumoto K, Abe I, Fujiki Y (2000) Molecular anatomy of the peroxin Pex12p: ring finger domain is essential for Pex12p function and interacts with the peroxisome-targeting signal type 1-receptor Pex5p and a ring peroxin, Pex10p. J Biol Chem 275:25700–25710
Osumi T, Imamura A, Tsukamoto T, Fujiwara C, Hashiguchi N, Shimozawa N, Suzuki Y, Kondo N (2000) Temperature sensitivity in peroxisome assembly processes characterizes milder forms of peroxisome biogenesis disorders. Cell Biochem Biophys 32:165–170
Poll-The BT, Gootjes J, Duran M, de Klerk JB, Wenniger-Prick LJ, Admiraal RJ, Waterham HR, Wanders RJ, Barth PG (2004) Peroxisome biogenesis disorders with prolonged survival: phenotypic expression in a cohort of 31 patients. Am J Med Genet A 126:333–338
Raas-Rothschild A, Wanders RJ, Mooijer PA, Gootjes J, Waterham HR, Gutman A, Suzuki Y, Shimozawa N, Kondo N, Eshel G, Espeel M, Roels F, Korman SH (2002) A PEX6-defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents. Am J Hum Genet 70:1062–1068
Reuber BE, Germain-Lee E, Collins CS, Morrell JC, Ameritunga R, Moser HW, Valle D, Gould SJ (1997) Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet 17:445–448
Roels F, Saudubray JM, Giros M, Mandel H, Eyskens F, Saracibar N, Atares PB, Prats JM, De Prest B, De Preter K, Pineda M, Krystkowiak P, Gootjes J, Wanders RJ, Espeel M, Poll-The BT (2003) Peroxisome mosaics. Adv Exp Med Biol 544:97–106
Sacksteder KA, Jones JM, South ST, Li X, Liu Y, Gould SJ (2000) PEX19 binds multiple peroxisomal membrane proteins, is predominantly cytoplasmic, and is required for peroxisome membrane synthesis. J Cell Biol 148:931–944
Schutgens RB, Schrakamp G, Wanders RJ, Heymans HS, Tager JM, van den BH (1989) Prenatal and perinatal diagnosis of peroxisomal disorders. J Inherit Metab Dis 12(Suppl 1):118–134
Steinberg S, Chen L, Wei L, Moser A, Moser H, Cutting G, Braverman N (2004) The PEX Gene Screen: molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum. Mol Genet Metab 83:252–263
Steinberg S, Katsanis S, Moser A, Cutting G (2005) Biochemical analysis of cultured chorionic villi for the prenatal diagnosis of peroxisomal disorders: biochemical thresholds and molecular sensitivity for maternal cell contamination detection. J Med Genet 42:38–44
Valianpour F, Selhorst JJ, van Lint LE, van Gennip AH, Wanders RJ, Kemp S (2003) Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab 79:189–196
Vallance H, Applegarth D (1994) An improved method for quantification of very long chain fatty acids in plasma. Clin Biochem 27:183–186
Walter C, Gootjes J, Mooijer PA, Portsteffen H, Klein C, Waterham HR, Barth PG, Epplen JT, Kunau WH, Wanders RJ, Dodt G (2001) Disorders of peroxisome biogenesis due to mutations in PEX1: phenotypes and PEX1 protein levels. Am J Hum Genet 69:35–48
Wanders RJ (2004) Metabolic and molecular basis of peroxisomal disorders: a review. Am J Med Genet A 126:355–375
Wanders RJ, Dekker C, Ofman R, Schutgens RB, Mooijer P (1995a) Immunoblot analysis of peroxisomal proteins in liver and fibroblasts from patients. J Inherit Metab Dis 18(Suppl 1):101–112
Wanders RJ, Denis S, Ruiter JP, Schutgens RB, van Roermund CW, Jacobs BS (1995b) Measurement of peroxisomal fatty acid beta-oxidation in cultured human skin fibroblasts. J Inherit Metab Dis 18(Suppl 1):113–124
Wanders RJ, Schutgens RB, van den Bosch H, Tager JM, Kleijer WJ (1991) Prenatal diagnosis of inborn errors in peroxisomal beta-oxidation. Prenat Diagn 11:253–261
Wanders RJ, van Roermund CW (1993) Studies on phytanic acid alpha-oxidation in rat liver and cultured human skin fibroblasts. Biochim Biophys Acta 1167:345–350
Wanders RJ, Waterham HR (2005) Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin Genet 67:107–133
Wanders RJ, Wiemer EA, Brul S, Schutgens RB, van den Bosch H, Tager JM (1989) Prenatal diagnosis of Zellweger syndrome by direct visualization of peroxisomes in chorionic villus fibroblasts by immunofluorescence microscopy. J Inherit Metab Dis 12 (Suppl 2):301–4
Yajima S, Suzuki Y, Shimozawa N, Yamaguchi S, Orii T, Fujiki Y, Osumi T, Hashimoto T, Moser HW (1992) Complementation study of peroxisome-deficient disorders by immunofluorescence staining and characterization of fused cells. Hum Genet 88:491–499
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zeharia, A., Ebberink, M.S., Wanders, R.J.A. et al. A novel PEX12 mutation identified as the cause of a peroxisomal biogenesis disorder with mild clinical phenotype, mild biochemical abnormalities in fibroblasts and a mosaic catalase immunofluorescence pattern, even at 40 °C. J Hum Genet 52, 599–606 (2007). https://doi.org/10.1007/s10038-007-0157-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0157-y
Keywords
This article is cited by
-
A founder mutation in PEX12 among Egyptian patients in peroxisomal biogenesis disorder
Neurological Sciences (2021)
-
Mild Zellweger syndrome due to a novel PEX6 mutation: correlation between clinical phenotype and in silico prediction of variant pathogenicity
Journal of Applied Genetics (2017)
-
Zellweger spectrum disorders: clinical overview and management approach
Orphanet Journal of Rare Diseases (2015)
-
Rational diagnostic strategy for Zellweger syndrome spectrum patients
European Journal of Human Genetics (2009)
