
Abstract
Rett syndrome (RTT) is a progressive neurodevelopmental disorder that is caused by mutations in the X-linked methyl-CpG-binding protein2 (MECP2) gene. In this study, the MECP2 sequences in 121 unrelated Chinese patients with classical or atypical RTT were screened for deletions and mutations. In all, we identified 45 different MECP2 mutations in 102 of these RTT patients. The p. T158M mutation (15.7%) was the most common, followed in order of frequency by p. R168X (11.8%), p. R133C (6.9%), p. R270X (6.9%), p. G269fs (6.9%), p. R255X (4.9%), and p. R306C (3.9%). In addition, we identified five novel MECP2 mutations: three missense (p. K305E, p. V122M, p. A358T), one insertion (c.45-46insGGAGGA), and one 22 bp deletion (c.881-902del22). Large deletions represented 10.5% of all identified MECP2 mutations. Conversely, mutations in exon 1 appeared to be rare (0.9%). The remaining cases without MECP2 mutations were screened for the cyclin-dependent kinase-like 5 (CDKL5) gene using denaturing high-performance liquid chromatography (DHPLC). One synonymous mutation (p. I72I) was found in exon 5, suggesting that CDKL5 is a rare cause of RTT. The overall MECP2 mutation detection rate for this patient series was 84.3:87.9% in 107 classical RTT cases and 57.1% in 14 atypical RTT cases. Moreover, there were two patients with homozygous mutations and normal female karyotypes. However, we did not pinpoint a significant relationship between genotype and phenotype in these cases.
Similar content being viewed by others


Introduction
Rett syndrome (RTT, MIM 312750) is a progressive neurodevelopmental disorder that affects females almost exclusively, with an estimated prevalence of approximately 1 in 10,000–15,000 females. RTT is often considered an X-linked dominant condition with male lethality (Hagberg 1985) characterized by a progressive loss of intellectual function, fine gross motor skills, and communicative abilities; deceleration of head growth; and the development of stereotypic hand movements, all occurring after a period of normal development.
In 1999, Amir et al. (1999) determined that RTT is caused by mutations in the X-linked methyl-CpG-binding protein2 ( MECP2) gene on chromosome Xq28. Prior to the identification of the MECP2B isoform, mutation detection efforts focused on using polymerase-chain-reaction (PCR)-based approaches to screen exons 2, 3, and 4. The recent identification of yet another MeCP2 isoform, MeCP2A, has enhanced speculation that some individuals may have mutations in exon 1 (Mnatzakanian et al. 2004).
To date, mutations have been identified in the MECP2 sequences of approximately 80% of all RTT patients; the remaining 20% may possess noncoding regions of this gene, or they may harbor a second RTT-inducing gene or locus. Several reports have identified large gene deletions in RTT patients that escaped detection by PCR-based screening strategies (Laccone et al. 2004; Schollen et al. 2003). Others have identified mutations in a novel MeCP2 isoform and in CDKL5 (Archer et al. 2006a, b; Evans et al. 2005a; Kriaucionis et al. 2004; Mari et al. 2005; Scala et al. 2005; Tao et al. 2004; Weaving et al. 2004). This study reports the results of the mutation analysis of MECP2 and CDKL5 in 121 unrelated Chinese patients with classic or atypical RTT, conducted to obtain a genotypic representation of the mutational spectrum in this population.
Materials and methods
Patients
A total of 121 sporadic RTT patient cases, consisting of 107 classic and 14 atypical patients, were involved in this study. The age of RTT onset ranged from 3–36 months, and the oldest patient living with RTT was 24 years of age. The cases, all female, were distributed across 27 Chinese provinces. All patients fulfilled the classic or atypical diagnostic criteria proposed by The Rett Syndrome Diagnostic Criteria Work Group (Hagberg et al. 2002) and in 14 atypical patients, six had preserved speech variant, two early onset seizures, one forme fruste, and five were unclassified. Informed parental consent was obtained for all patients.
Mutation detection
Genomic DNA was extracted using standard procedures from the peripheral blood leukocytes of patients with RTT (Miller et al. 1988).
DNA direct sequencing
The PCR method was used to amplify the four MECP2 exons by using published primers (Amir et al. 1999; Mnatzakanian et al. 2004). If no mutations were identified after screening exons 2–4, then exon 1 was screened. The final volume of the PCR was 25 μl, consisting of 50 ng of DNA, 0.005 mM of each primer pair, 2.5 mM dNTPs, 1.5 mM MgCl2, 1× reaction buffer, and 1U Taq DNA polymerase (Promega or Invitrogen). To amplify exon 1, the 500 mM of betain and 50% dimethylsulfoxide were added to the PCR system, which was different with exons 2, 3, and 4. The PCR conditions used were as follows: initial denaturation at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 52–62°C for 30 s, initial extension at 72°C for 30–60 s, and final extension at 72°C for 10 min. The PCR products were purified using a QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA). The purified products were then sequenced using a BigDye Terminator Cycle Sequencing Kit and an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). The sequencing primers were the same as those used for the PCR amplification. Mutation analyses were performed using the normal, non-RTT human genomic MECP2 sequence as a reference (GenBank accession No. AF030876).
To confirm that alterations found in this work were novel mutations and not polymorphisms, 100 normal control alleles were examined directly using PCR-restriction fragment-length polymorphism (RFLP) techniques. If the mutation was not associated with a RFLP, then DNA sequencing or denaturing high-performance liquid chromatography (DHPLC) were conducted. All variations were verified by sequencing with the forward and reverse primers.
Multiplex ligation-dependent probe amplification
If no mutation was identified with PCR in the MECP2 sequences of RTT patients, then we used multiplex ligation-dependent probe amplification (MLPA) kit P015C (MRC-Holland, Amsterdam, The Netherlands) to screen for large deletions. The MLPA reactions were performed following the instructions provided by the manufacturer. Finally, the PCR products were separated by capillary electrophoresis using an ABI 3100 automated sequencer and size standards (Perkin Elmer Applied Biosystems, Foster City, CA USA). We used Genescan analysis software (version 3.7) and Genotype software (version 3.6) to analyze these data, which we then exported to a Microsoft Excel spreadsheet. The resulting values were approximately 1.0 for every wild type peak, 0.5 for heterozygous deletions, and 1.5 for heterozygous duplications.
Long-range PCR and sequencing
The large deletions were verified using long-range PCR and direct sequencing methods. The PCR amplifications were performed, as described by Amir et al. (1999) with the following modifications. Exons 3 and 4 were amplified using a pair of primers, comprised of upstream (exon 3-1F: 5′-GTTCCCCCCGACCCCACCCT-3′, which was localized within intron 2) and downstream (exon 4-4R: 5′-CTCCCTCCCCTCGGTGTTTG-3′) primers. This amplification generated a fragment of 2,171 bp. The PCR volume was 25 μl with GC buffer I, 2.5 mM dNTP, 0.005 mM of each primer, 100 ng genomic DNA, and 2.5 U of TaKaRa LA Taq (TaKaRa, Tokyo, Japan). An initial denaturing step of 94°C for 2 min was performed, followed by 35 cycles at 94°C for 10 s and 68°C for 150 s. All reactions were terminated by a final elongation step at 72°C for 10 min. The PCR products were sequenced, as described above.
CDKL5 gene mutation screening by DHPLC
If no mutations were identified in the MECP2 sequence isolated from the RTT patients using general PCR and MLPA methods, then the coding region of CDKL5 was screened by DHPLC. The PCR amplifications were conducted, as described above. The melting temperatures (Tms) for all primer pairs and a full list of DHPLC-run temperatures are listed in Table 1. The DHPLC was performed on a WAVE Nucleic Acid Fragment Analysis System HSM (Transgenomic, Crewe, UK). The PCR products displaying abnormal chromatographic profiles on the DHPLC analysis were sequenced directly.
X-chromosome inactivation
The analysis of X-chromosome inactivation (XCI) was performed in the RTT patients, as well as the mothers of certain patients, as described and recommended by Allen et al. (1992). In brief, the lymphocyte genomic DNA was digested with the methylation-sensitive enzyme Hpa II prior to the PCR amplification of the polymorphic CAG repeat in the androgen receptor (AR) gene. The amplicons were sequenced with an ABI 3100 automated sequencer (Applied Biosystems), and the resulting sequences were analyzed with GeneScan analysis software. The peak areas were compared before and after the Hpa II digestions (data not shown).
Results
In total, 45 different MECP2 mutations were identified in 102 of the 121 diagnosed sporadic female patients presenting with classical or atypical RTT (Table 2). One mutation was found in exon 5 of CDKL5. The p. A358T mutation was also observed in the tested maternal DNA. The other mutations were not found in the parents, which indicated that these mutations arose de novo.
MECP2 mutation detection by direct sequence analysis
Mutations in MECP2 were detected in 87.9% (94/107) of the patients presenting with classic RTT and in 57.1% (8/14) of those with atypical RTT. These aberrations consisted of 71 point mutations, 17 microdeletions, 11 large deletions, 2 insertions, and 1 splicing defect. Most of the variants were missense mutations, accounting for 43.1% (44/102), followed in order of frequency by nonsense mutations 26.5% (27/102), and frame shift mutations 17.6% (18/102). The p.T158M (c.473C > T, 15.7%, 16/102) was the most common of the MECP2 mutations, followed by p. R168X (c.502C > T, 11.8%, 12/102), p. R133C (c.397C > T, 6.9%, 7/102), p. R270X (c.808C > T, 6.9%, 7/102), c. 806delG (6.9%, 7/102), p. R255X (c.763C > T, 4.9%, 5/102), and p. R306C (c.916C > T, 3.9%, 4/102).
We also identified five novel mutations, three of which were point mutations. The first, a G > A transition at the cDNA position 364 (c.364G > A, Fig. 1a), lead to a novel methionine to valine substitution at residue 122 (p. V122M). The second novel mutation was an A > G transition at cDNA position 916 (c.916 G > A, Fig. 1b), leading to a glutamate to lysine substitution at residue 305 (p. K305E). Moreover, this mutation eliminated a Hae II restriction site. The third novel mutation was a G > A transition at cDNA position 1076 (c.1076G > A, Fig. 1c), which caused a substitution of alanine with a threonine at position 358 and generated an Hpy188 III restriction site. Using RFLP analysis and these two enzymes, we tested the parental samples and the normal female controls to distinguish between the wild-type and mutant alleles. The first and second mutations were deemed de novo, as they were not detectable in the parents and 100 normal female controls. The p. A358T mutation was present in the patient’s mother and absent in all 100 control alleles. A repeat GGAGGA insertion was detected at cDNA position 45–46 in exon 1 of MECP2 (Fig. 1d). Additionally, one novel 22-bp microdeletion at cDNA position 881–902 in MECP2 exon 4 resulted in a frameshift (Fig. 1e).

Nucleotide sequences of five novel mutations of X-linked methyl-CpG-binding protein2 (MECP2) gene in patients with Rett syndrome (RTT). The blackened arrow indicates the affected nucleotide. Deleted or inserted bases are indicated above. a c.364G > A (p.V122M); b c.916G > A (p.K305E); c c.1076G > A (p.A358T); d c.45_46insGGAGGA (ATG from exon1); e c.881-902del22
Notably, there were two RTT patients with mutations (p. P127L, p. T158M) in an apparently homozygous condition. The two mutations were not found in the parental samples.
MECP2 mutation detection by MLPA analysis and long-range PCR
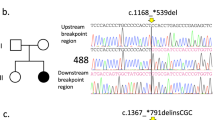
The 27 RTT patients who did not exhibit any mutations in MECP2 after the PCR screening were examined with MLPA. Using this alternate method, we identified large single-exon and multi-exon deletions in 9.1% (11/121) of all MECP2 mutations. More than 90% of these deletions involved exons 3 and/or 4. Seven of the 11 large deletions involved two exons: six in exons 3 and 4, and one in exons 1 and 2 of MECP2. Four deletions involved only part of exon 4. In one case, the flanking IRAK1 gene was deleted along with exons 3 and 4 of MECP2 (Pan et al. 2006, submitted). The breakpoints were identified in two large deletions in exon 4 using long-range PCR followed by direct sequencing of the amplicons. In one case, the upstream breakpoint was located at cDNA position 905 whereas the downstream breakpoint was located at cDNA position 1,168, resulting in the deletion of 234 bp. In the other case, one of the breakpoints was located within the deletion-prone region (DPR) whereas the downstream breakpoint was located at cDNA position 1,047, resulting in a deletion of 152 bp.
CDKL5 mutation detection by DHPLC analysis
In the remaining 16 RTT patients without MECP2 mutations, a synonymous mutation (p. I72I) was detected in exon 5 of CDKL5. We were unable to sample and test the parental DNA, but we did not detect this mutation in 100 of the normal controls, as determined with DHPLC.
Discussion
MECP2 mutation detection
Mutations in MECP2 as causative factors for RTT have been reported worldwide, for example, in Japan, the United States, and France (Amano et al. 2000; Amir et al. 1999; Philippe et al. 2006). The mutations are distributed along the entire MECP2 gene and comprise all mutation categories. Mutation distribution may be considered tripartite. Missense mutations are clustered prevalently in the methyl-CpG-binding domain (MBD). Nonsense mutations are more scattered but tend to be located between the MBD and transcription repression domain (TRD) or within the TRD. This preferential location may reflect the functional importance of the domains or might be the consequence of a marked sequence-specific hypermutability region (Miltenberger-Miltenyi and Laccone 2003).
The study presented herein represents mutation data from a Chinese population consisting of 121 sporadic RTT patients, 107 and 14 of whom presented with classic or atypical RTT, respectively. Mutations were detected in 87.9% (94/107) of patients with classic RTT, which is consistent with the estimated reports. Roughly 57.1% (8/14) of the patients with atypical RTT possessed mutations in their MECP2 sequences. Recent reports described a range of 65–79% for the mutation detection rate among patients with atypical RTT (Smeets et al. 2003, 2005). Kanmmoun et al. recommended selecting well-defined clinical criteria while searching for MECP2 mutations, as all individual variables did not exhibit similar weights (Kammoun et al. 2004). Thus, we hypothesized that the different mutation frequencies of atypical RTT were correlated with different clinical information about atypical RTT. The total mutation frequency among all RTT patients was 84.3% (102/121).
In our study, we determined that 88/121 (72.7%) of the Chinese RTT alleles were mutated in exon 4 (c.378_1461 of MECP2A), which encodes for a portion of MBD and TRD. A few mutations (occurring in five cases) were located in exon 3 (c.27_377) whereas no mutation was identified in exon 2 (c.1_26). Mutations located in MECP2B exon 1 are rare, as they represent just 0.9% of all MECP2 mutations. We report here five novel MECP2 mutations: one in exons 1 and 3 and three in exon 4. The most frequent point mutations were determined to be missense (41.1%), which tended to cluster in the MBD (aa 90–174, 35/43, 81.4%) and in the TRD (aa 219–322, 6/43, 14.0%). Nonsense mutations represented 26.7% of all examined MECP2 mutations and tended to cluster in the MBD and TRD. Nearly half (12/27, 44.4%) of the nonsense mutations deleted a portion of the nuclear localization signal (NLS) (aa 267–283) or the MBD and are not likely to encode an actively transported nuclear protein, and they may reduce the DNA binding capacity, respectively. The seven most frequently observed and reported MECP2 mutations represent 56.9% (58/102) of all of the identified mutations in our study. Moreover, in some reports, the most common mutation is p. R168X (Cheadle et al. 2000; Dragich et al. 2000; Wan et al. 1999). However, the p. T158M was a predominant finding herein, which is consistent with our previous report (Pan et al. 2002) and with reports on cohorts of American (Buyse et al. 2000), British-Italian (Vacca et al. 2001), Japanese (Fukuda et al. 2005), and Korean descent (Chae et al. 2002).
The DPR, which represents a highly repetitive region in MECP2 exon 4, is located in the region that is 3′ to the TRD, where many intragenic deletion breakpoints occur (C-terminal deletions) (Laccone et al. 2004). In our study, 16 different or complex deletions were located in the DPR, representing 11.0% (16/121) of the MECP2 mutations. These deletions were similar to those recently isolated by Laccone et al. (2004). Recombination between the DPR and a second highly repetitive region located in intron 2 may be responsible for mediating the commonly identified large deletions encompassing exons 3 and 4. It is also likely that there are recombinogenic repetitive elements in the 3′UTR that give rise to the large intragenic deletions found in exon 4 (Archer et al. 2006a, b). One previous report suggested that truncated MeCP2 proteins may degrade faster in vivo, which would contribute to their loss of function (Yusufzai and Wolffe 2000). The other report (Chandler et al. 1999), however, showed that the deletion of the 63 amino acids at the C-terminus of MeCP2 impairs its DNA binding capacity during the transcription regulation process. Each patient in 11 RTT cases presenting with large MECP2 deletions were missing one or two exons. The four patients who had deletions in exon 4 did not experience the characteristic seizures and scoliosis whereas the six patients with deletions in exons 3 and 4 endured seizures, complete loss of hand function, no language ability, and accompanying scoliosis (Pan et al. 2006, submitted). One previous study suggested that RTT phenotype is more likely to be complete as the child grows up (Kammoun et al. 2004). In our study, the majority of patients were younger than 10 years. Thus, we intend to conduct follow-up analyses on these patients to augment our sample population. To our knowledge, only six cases, including those in our study, presented with deletions in exons 1 and/or 2 (Archer et al. 2006a, b; Erlandson et al. 2003). Therefore, exon 1 deletions or mutations are rare.
One patient had a novel MECP2 missense mutation (c.1076G > A, p. A358T) that was inherited maternally, although the mother possessed a normal phenotype. The XCI demonstrated a preferential inactivation of the paternal allele, with a frequency of 82%. The maternal allele XCI analysis yielded no information (Fig. 2). It is possible that the mother had gonadal mosaicisms or she was spared from the disease by a favorable X-inactivation event, as reported previously (Amir et al. 1999; Wan et al. 1999). We also found a 6-bp insertion in the [GGA]5trinucleotide repeats in exon 1 of the patients who did not have mutations in exons 2, 3, or 4 of MECP2, thus the patient has a classic manifestation. This patient lost her speaking ability at 18 months, had stereotypical hand movements, and did not feed herself after the onset of developmental delay. We suggested that a 45–46insGGAGGA was a potential pathogenic mutation. Taken together, these findings, including our 16 cases with the earlier published finding from exon 1, revealed that nine exon 1 mutations have been detected among 245 mutation-negative RTT patients (Amir et al. 2005; Bartholdi et al. 2006; Evans et al. 2005b; Laccone 2004; Ravn et al. 2005; Saxena et al. 2006). At a practical level, the prevalence of exon 1 mutations estimated is 3.7% among mutation-negative RTT cases whereas the overall contribution of exon 1 mutation to the occurrence of RTT is less than 1%. Our findings confirm the emerging consensus that mutations in exon 1 are responsible for only a minority of RTT cases.

A358T X-inactivation pattern of the patient and her mother. The upper automated sequencer traces show two alleles of the androgen receptor of the patient; the lower automated sequencer traces show the alleles of the androgen receptor of the patient’s mother; the left is the trace of undigested by restriction enzymes HpaII, the right is the trace of digested by HpaII
A majority of mutations identified herein were heterozygous; however, two that we identified were homozygous. p. P322L and p. T158M (patients 119 and 120, respectively) were not present in their parents, which implicated the existence of one of these deletions on the parental alleles. One other study also identified this condition (Schollen et al. 2003). It is recommended that future MECP2 mutation screenings attempt to avoid overlooking similar mutations in RTT patients. Nearly 77.7% (94/121) of the classic and atypical RTT individuals showed changes within the coding region of exon 4. This finding suggests that an initial analysis of exon 4 would provide the most efficient approach in a mutation protocol.
CDKL5 mutation analysis
A small handful of mutations in CDKL5 has been identified recently in patients with an early seizure phenotype of atypical RTT (Archer et al. 2006a, b; Evans et al. 2005a; Mari et al. 2005; Scala et al. 2005; Tao et al. 2004; Weaving et al. 2004). To date, 15 different CDKL5 mutations have been identified in 15 atypical RTT patients with early onset seizure disorder. In an attempt to identify the mutations in the remaining 16 RTT patients without a defined MECP2 alteration, we used DHPLC to screen the coding region of CDKL5. From these screenings, we identified one case with a synonymous mutation (c.216T > A, p. I72I) in exon 5. Unfortunately, the parental samples were not available for analysis; however, we did not detect the mutation in 100 of the normal control alleles. This finding suggests that this mutation is related to the RTT. This patient manifested an atypical RTT, could speak a single word at 8 months, lost her speaking capability at 12 months, and then experienced spasms. She presented with stereotypical hand rubbing and did not get scoop accompanied with grind. Presently, she can walk, has regained some hand function, but is autistic.
In conclusion, mutations in exon 4 of MECP2 appear to be the most frequent among those found in Chinese RTT patients. The mutation in MECP2 exon 1 and in CDKL5 appears to be rare. Our data suggest that escape from PCR detection is a fairly common event, especially among the larger deletions, and that routine mutation screening in MECP2 should include quantitative analysis of the MECP2, preferably with MLPA. We have found that MLPA, as a complement to DNA sequencing, is a useful tool for detecting whole-exon deletions.
References
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JM (1992) Methylation of HpaII and HhaII sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51:1229–1239
Amano K, Nomura Y, Segawa M, Yamakawa K (2000) Mutational analysis of the MECP2 gene in Japanese patients with Rett syndrome. J Hum Genet 45:231–236
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutation in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188
Amir RE, Fang P, Yu Z, Glaze DG, Percy AK, Zoghbi HY, Roa BB, Van den Veyver IB (2005) Mutations in exon 1 of MECP2 are a rare cause of Rett syndrome. J Med Genet 42:e15
Archer HL, Evans JC, Edwards S, Colley J, Newbury-Ecob R, O’callaghan F, Huyton M, O’Regan M, Tolmie J, Sampson J, Clarke AJ, Osborne J (2006a) CDKL5 mutations cause infantile spasms, early onset seizures and severe mental retardation in female patients. J Med Genet 12 Apr (published online)
Archer HL, Whatley SD, Evans JC, Ravine D, Huppke P, Kerr A, Bunyan D, Kerr B, Sweeney E, Davies SJ, Reardon W, Horn J, MacDermot KD, Smith RA, Magee A, Donaldson A, Crow Y, Hermon G, Miedzybrodzka Z, Cooper DN, Lazarou L, Butler R, Sampson J, Pilz DT, Laccone F, Clarke AJ (2006b) Gross rearrangements of the MECP2 gene are found in both classical and atypical Rett Syndrome. J Med Genet 43:451–456
Bartholdi D, Klein A, Weissert M, Koenig N, Baumer A, Boltshauser E, Schinzel A, Berger W, Matyas G (2006) Clinical profiles of four patients with Rett syndrome carrying a novel exon 1 mutation or genomic rearrangement in the MECP2 gene. Clin Genet 69:319–326
Buyse IM, Fang P, Hoon KT, Amir RE, Zoghbi HY, Roa BB (2000) Diagnostic testing for Rett syndrome by dHPLC and direct sequencing analysis of the MECP2 gene: identification of several novel mutations and polymorphisms. Am J Hum Genet 67:1428–1436
Chae JH, Hwang YS, Kim KJ (2002) Mutation analysis of MECP2 and clinical characterization in Korean patients with Rett syndrome. J Child Neurol 17:33–36
Chandler SP, Guschin D, Landsberger N, Wolffe AP (1999) The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry 38:7008–7018
Cheadle JP, Gill H, Fleming N, Maynard J, Kerr A, Leonard H, Krawczak M, Cooper DN, Lynch S, Thomas N, Hughes H, Hulten M, Ravine D, Sampson JR, Clarke A (2000) Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Hum Mol Genet 9:1119–1129
Dragich J, Houwink-Manville I, Schanen C (2000) Rett syndrome: a surprising result of mutation in MECP2. Hum Mol Genet 9:2365–2375
Erlandson A, Samuelsson L, Hagerg B, Kyllerman M, Vujic M, Vujic M, Wahlstrom J (2003) Multiplex ligation-dependent probe amplification (MLPA) detects large deletions in the MECP2 gene of Swedish Rett syndrome patients. Genet Test 7:329–332
Evans JC, Archer HL, Colley JP, Ravn K, Nielsen JB, Kerr A, Williams E, Christodouliu J, Gecz J, Jardine PE, Wright MJ, Pilz DT, Lazarou L, Cooper DN, Sampson JR, Butler R, Whatley SD, Clarke AJ (2005a) Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet 13:1113–1120
Evans JC, Archer HL, Whatley SD, Kerr A, Clarke A, Butler R (2005b) Variation in exon 1 coding region and promoter of MECP2 in Rett syndrome and controls. Eur J Hum Genet 13:124–126
Fukuda T, Yamashita Y, Nagamitsu S, Miyamoto K, Jin JJ, Ohmori I, Ohtsuka Y, Kuwajima K, Endo S, Iwai T, Yamagata H, Tabara Y, Miki T, Matsuishi T, Kondo I (2005) Methyl-CpG binding protein 2 gene (MECP2) variations in Japanese patients with Rett syndrome: pathological mutations and polymorphisms. Brain Dev 27:211–217
Hagberg B (1985) Rett’ syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr Scand 74:405–408
Hagberg B, Hanefeld F, Percy A, Skjeldal O (2002) An update on clinically applicable diagnostic criteria in Rett syndrome. Eur J Paediatric Neurol 6:293–297
Kammoun F, De Roux N, Boespflug-Tanguy O, Vallee L, Seng R, Tardieu M, Landrieu P (2004) Screening of MECP2 coding sequence in patients with phenotypes of decreasing likelihood for Rett syndrome: a cohort of 171 cases. J Med Genet 41:e85
Kriaucionis S, Bird A (2004) The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res 32:1818–1823
Laccone F, Junemann I, Whatley S, Morgan R, Butler R, Huppke P, Ravine D (2004) Large deletions of the MECP2 detected by gene dosage analysis in patients with Rett syndrome. Hum Mutat 23:234–244
Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, Scala E, Longo I, Grosso S, Pescucci C, Ariani F, Hayek G, Balestri P, Bergo A, Badaracco G, Zappella M, Broccoli V, Renieri A, Kilstrup-Nielsen C, Landsberger N (2005) CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet 14:1935–1946
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Miltenberger-Miltenyi G, Laccone F (2003) Mutations and Polymorphisms in the Human Methyl- CpG-binding Protein MeCP 2. Hum Mutat 22:107–115
Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJ, Jones JR, Scherer SW, Schanen NC, Friez MJ, Vincent JB, Minassian BA (2004) A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet 36:339–341
Pan H, Wang YP, Bao XH, Meng HD, Zhang Y, Wu XR, Shen Y (2002) MECP2 gene mutation analysis in Chinese patients with syndrome. Eur J Hum Genet 10:484–486
Philippe C, Villard L, De Roux N, Raynaud M, Bonnefond JP, Pasquier L, Lesca G, Mancini J, Jonveaux P, Moncla A, Chelly J, Bienvenu T (2006) Spectrum and distribution of MECP2 mutations in 424 Rett syndrome patients: a molecular update. Eur J Med Genet 49:9–18
Ravn K, Nielsen J, Schwarta M (2005) Mutations found within exon 1 of MECP2 in Danish patients with Rett syndrome. Clin Genet 67:532–533
Saxena A, De lagarde D, Leonard H, Williamson SL, Vasudevan V, Christodoulou J, Thompson E, MacLeod P, Ravine D (2006) Lost in translation: translational interference from a recurrent mutation in exon 1 of MECP2. J Med Genet 43:470–477
Scala E, Ariani F, Mari F, Caselli R, Pescucci C, Longo I, Meloni I, Giachino D, Bruttini M, Hayek G, Zapella M, Renieri A (2005) STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet 42:103–107
Schollen E, Smeets E, Deflem E, Fryns JP, Matthijs G (2003) Gross rearrangements in the MECP2 gene in three patients with Rett syndrome: implications for routine diagnosis of Rett syndrome. Hum Mutat 22:116–120
Smeets E, Schollen E, Moog U, Matthijs G, Herbergs J, Smeets H, Curfs L, Schrander-Stumpel C, Fryns JP (2003) Rett syndrome in adolescent and adult females: Clinical and molecular genetic findings. Am J Med Genet 122:227–233
Smeets E, Terhal P, Casaer P, Peters A, Midro A, Schollen E, Van Roozendaal K, Moog U, Matthijs G, Herbergs J, Smeets H, Curfs L, Schrander-Stumpel C, Fryns JP (2005) Rett syndrome in females with CTS hot spot deletions: a disorder profile. Am J Med Genet 132:117–120
Tao J, Van Esch H, Hagedorn-Greiwe M, Hoffman K, Moser B, Raynaud M, Sperner J, Fryns JP, Schwinger E, Gecz J, Ropers HH, Kalscheuer VM (2004) Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet 75:1149–1154
Vacca M, Filippini F, Budillon A, Rossi V, Della Ragione F, De Bonis ML, Mercadante G, Manzati E, Gualandi F, Bigoni S, Trabanelli C, Pini G, Calzolari E, Ferlini A, Meloni I, Hayek G, Zappella M, Renieri A, D’Urso M, D’Esposito M, Macdonald F, Kerr A, Dhanjal S, Hulten M (2001) MECP2 gene mutation analysis in the British and Italian Rett Syndrome patients: hot spot map of the most recurrent mutation and bioinformatics analysis of a new MECP2 conserved region. Brain Dev 23:S246-S250
Wan M, Lee SS, Zhang X, Houwink-Manville I, Song HR, Amir RE, Budden S, Naidu S, Pereira JL, Lo IF, Zoghbi HY, Schanen NC, Francke U (1999) Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am J Hum Genet 65:1520–1529
Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H, Evans J, Clarke A, Pelka GJ, Tam pp, Watson C, Lahooti H, Ellaway CJ, Bennetts B, Leonard H, Gecz J (2004) Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 75:1079–1093
Yusufzai TM, Wolffe AP (2000) Functional consequences of Rett syndrome mutations on human MeCP2. Nucleic Acids Res 28:4172–4179
Acknowledgments
We thank all the families taking part in this study. This work was supported by the National Natural Science Foundation of China (30271374).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Li, Mr., Pan, H., Bao, XH. et al. MECP2 and CDKL5 gene mutation analysis in Chinese patients with Rett syndrome. J Hum Genet 52, 38–47 (2007). https://doi.org/10.1007/s10038-006-0079-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0079-0
Keywords
This article is cited by
-
Variation spectrum of MECP2 in Korean patients with Rett and Rett-like syndrome: a literature review and reevaluation of variants based on the ClinGen guideline
Journal of Human Genetics (2022)
-
Variant Profile of MECP2 Gene in Sri Lankan Patients with Rett Syndrome
Journal of Autism and Developmental Disorders (2020)
-
The presence of two rare genomic syndromes, 1q21 deletion and Xq28 duplication, segregating independently in a family with intellectual disability
Molecular Cytogenetics (2016)
-
Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes
neurogenetics (2009)
