
Abstract
Proteolytic stress, resulting from the intracellular accumulation of misfolded or aggregated proteins, which exceed the capacity of the ubiquitin–proteasome system to degrade them, plays a relevant role in neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s chorea. Most of toxic protein aggregates are characterised by the presence of isopeptide bonds (cross-links) catalysed by transglutaminase activity; further, several disease-specific proteins—tau, amyloid-beta, alpha-synuclein, huntingtin—are in vitro and/or in vivo substrates of transglutaminase 2. These findings suggest an important role for transglutaminase 2-mediated cross-linking reactions in neurodegeneration. Therefore, the use of transglutaminase activity inhibitors could ameliorate neuronal cell death. New therapeutic perspectives also arise from the possibility to prevent or reduce protein aggregation by enhancing the activation of heat shock proteins, which have been shown to be potent suppressors of neurodegeneration in cell cultures/animal models. Interestingly, some heat shock proteins have been shown to be in vitro or in vivo cross-linked by transglutaminase 2. These observations seem to suggest that transglutaminase activity could be involved in the stabilization of intracellular protein aggregates by interfering with proteasomal degradation of misfolded proteins. Further studies are needed to validate leading hypotheses and to open new prospects for developing therapeutic tools.
Similar content being viewed by others


Introduction
A common feature of several neurodegenerative diseases of the central nervous system (CNS), such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington’s chorea (HD) is the accumulation of misfolded and/or aggregated proteins (Ross and Poirier 2004). The characterization of mechanisms underlying the formation of intracellular protein inclusions or extracellular protein aggregates might be important for the development of new therapeutic approaches of these diseases.
The current hypothesis, explaining how protein misfolding and aggregation might occur, would suggest that changes in the expression or post-translational modifications of a specific protein lead to misfolding. Then, alterations in the clearance pathways and/or the activity of molecular chaperones or other factors involved in the control of protein folding, may cause the formation of protein aggregates (Soto and Estrada 2008).
The relationships between protein aggregates and cell damage have been investigated using different in vitro and in vivo experimental models. Based on several observations, it is reasonable to hypothesize that the onset and/or progression of brain degeneration may be linked mechanistically to abnormal interactions between brain proteins, which lead to the assembly of the disease proteins into filaments, and the aggregation of these filaments within brain cells or in the extracellular space. The accumulation of large cytotoxic protein aggregates triggers proteolytic stress, which, in turn, causes the onset of programmed cell death in vulnerable brain regions (Mattson 2000). Aggregation-prone, disease-specific proteins and selectively affected brain regions in various neurodegenerative disorders are shown in Table 1.
Several in vitro and in vivo studies have shown that the enzyme activity of some transglutaminases (TGs) present in the CNS—namely, TG1, TG2, TG3, and TG5—might contribute to the formation of protein aggregates typical of AD, PD, HD, and other neurodegenerative diseases (Kim et al. 1999; Griffin et al. 2002; Muma 2007; Jeitner et al. 2009).
Transglutaminases and neurodegeneration
TGs are Ca2+-dependent enzymes that catalyse a variety of modifications of glutamine (Q) residues. The most common reaction is the inter- or intra-molecular protein transamidation (cross-linking), leading to the formation of stable soluble and insoluble polymers characterised by the presence of γ-glutamyl-ε-lysine (GGEL) isopeptide bonds (cross-links) between Q and K residues. Other functional reactions mediate polyamine incorporation and deamidation of Q residues in the absence of suitable amine acceptors (Griffin et al. 2002).
Under physiological conditions, the majority of cross-links are generated in the extracellular space where the Ca2+ concentration is high enough to stimulate TG activity. Intracellular Ca2+ concentration is usually lower than the extracellular one. Moreover, GTP and ATP act in cells as endogenous inhibitor of TGs (Griffin et al. 2002; Jeitner et al. 2009). Nevertheless, intracellular TG-catalysed reaction products can be detected in normal cells, especially those products related to polyamine incorporation into cell proteins (Jeitner et al. 2009).
In the brain, these modifications serve to either regulate enzyme activity or attach the TG substrates to biological matrices (Jeitner et al. 2009). However, only a small number of polyaminated proteins have been identified in the brain; thus, the effects of polyamination on nervous cells are poorly understood (Tucholski et al. 1999). Instead, TG cross-linking activity seems to play a key role in the protein aggregation process observed in most of neurodegenerative diseases. Post-mortem analysis of disease-affected brains demonstrated the co-localization of TG-mediated cross-links with aggregates of some disease-specific proteins, which are a typical feature of neurodegenerative diseases (Muma 2007).
Indeed, increased expressions of TGs have been reported in AD, PD, HD, and ALS (Muma 2007). Further, the activation of these enzymes in the above cited diseases is stimulated by perturbations in intracellular Ca2+ homeostasis, due to glutamate-mediated excitotoxicity and other toxic stimuli, as well as decreases in GTP concentrations following from losses in energy production (Jeitner et al. 2009).
TG2 and protein aggregation
Among various members of TGs family, TG2 is the most abundantly expressed in the human brain, being present in the amygdala, corpus callosum, cerebellum, and frontal cortex (Muma 2007). Other than in the cytosolic space, TG2 also localizes in the nucleus, mitochondria, at the cell surface, and in the extracellular matrix (ECM) (Griffin et al. 2002; Battaglia et al. 2007). Increased expression of TG2, accompanied by dysregulation of enzyme activity, has been observed in several neurodegenerative disorders (Muma 2007). Cell culture and animal experiments have shown that TG2 activation, other than by prolonged stimulation with excitatory amino acids and perturbation of calcium homeostasis, is triggered by various stress conditions, such as alterations of cell redox state, oxygen/glucose deprivation, inflammation, and extracellular matrix alterations (Ientile et al. 2002, 2004, 2007; Campisi et al. 2003, 2004; Caccamo et al. 2004). Interestingly, an important target of TG activity is the nuclear transcription factor-kappa B (NF-kappaB) (Lee et al. 2004), that is known to be involved in several conditions linked to neurodegeneration, such as excitotoxicity, oxidative stress, and inflammation (Mattson and Camandola 2001). TG-activated NF-kappaB, in turn, has been shown to sustain TG2 up-regulation in cell response to redox state alterations (Caccamo et al. 2005; Currò et al. 2009).
The involvement of TG2-mediated cross-linking reactions in the generation of protein aggregates is still controversial. Several in vitro studies have shown that protein transamidation by TG2 might contribute to the pathogenesis of neurodegenerative disorders, such as AD, PD and HD, by facilitating the formation and stabilization of disease-related protein oligomers, which result in toxicity for the nervous cells (Jeitner et al. 2009). Indeed, tau and amyloid-beta proteins, which typically aggregate in the intracellular neurofilaments and extracellular plaques, respectively, in AD-affected brains, have been reported as in vitro substrates of TG2 (Dudek and Johnson 1994; Miller and Johnson 1995). Moreover, tau polyamination can be observed in vivo (Tucholski et al. 1999).
However, a direct involvement of TG-mediated cross-linking in the generation of tau and amyloid-beta aggregates has not been proven, neither by immunohistochemical nor immunoenzymatic approaches (Singer et al. 2002; Bonelli et al. 2002), probably due to the poorly specific recognition of GGEL cross-links by commercially available antibodies (Johnson and LeShoure 2004).
Higher levels of TG2 proteins have been observed in dopamine neurons of brain substantia nigra, and in the cerebrospinal fluid (CSF) of PD patients compared with healthy subjects; also, the pattern of TG2 intracellular distribution in diseased dopamine neurons frequently overlaps that of alpha-synuclein, a PD-specific protein (Andringa et al. 2004).
Furthermore, cross-linking of alpha-synuclein as well as polyamination of several cell proteins are markedly enhanced in PD brains (Vermes et al. 2004; Jeitner et al. 2008).
Co-transfection studies in COS-7 cells, using mutant alpha-synuclein and TG2 recombinant vectors, demonstrated that the formation of alpha-synuclein oligomers was dependent upon enzymatically active TG2 since TG2 inactive mutants failed to induce aggregation. Also, the inhibition of TG activity by cystamine reduced the amount of alpha-synuclein aggregates (Junn et al. 2003). However, it has recently been demonstrated that TG2 binds equally to wild type and disease mutant alpha-synuclein variants, suggesting that the cross-linking of aggregation-prone proteins may prevent their progression into pathogenic species (Segers-Nolten et al. 2008).
The presence of elevated levels of GGEL cross-links in the CSF of HD patients support the hypothesis that in vivo TG activity is increased in HD brains (Jeitner et al. 2001). Indeed, TG-catalysed cross-links are also present in neuronal nuclei and co-localize with intracellular aggregates of huntingtin protein (Zainelli et al. 2003).
Interestingly, TG2 knockout provided beneficial effects in HD transgenic mice, as it produced significant increases in life span and improvement of motor dysfunction (Bailey and Johnson 2005). It has also been reported that TG activity inhibition by cystamine caused a reduction of huntingtin intracellular inclusions (Van Raamsdonk et al. 2005). However, results obtained in TG2−/− HD mice also suggested that the formation of inclusions might not depend on TG activity (Bailey and Johnson 2005). In the light of these findings, therapeutic approaches have been designed, and tested in animal models, to exploit the potential of several TG activity inhibitors which have displayed beneficial effects against protein aggregation in multiple biological models of neurodegenerative diseases (Siegel and Khosla 2007).
Heat shock proteins and proteolytic stress in neurodegenerative diseases
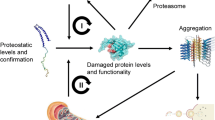
The accumulation of misfolded or aggregated proteins, which exceed the capacity of the ubiquitin–proteasome system to degrade them, usually triggers proteolytic stress within neuronal cells (Kopito and Ron 2000).
Given the relevant role played by proteolytic stress in neurodegeneration, a new classification as protein conformational diseases has been proposed for neurodegenerative disorders (Kopito and Ron 2000).
In this context, the role of heat shock proteins (HSPs) has attracted much attention, in that these molecular chaperones provide the first line of defence against misfolded or aggregation-prone proteins (Matthew and Morimoto 1998).
Constitutive HSPs assist the proper folding of newly synthesised proteins, ensure the maintenance of protein native conformation during stress conditions, and the recovery of misfolded proteins inside the cells. In addition, HSPs are involved in the trafficking of misfolded proteins to target organelles in order to facilitate the degradation by the ubiquitin–proteasome system or by the autophagic pathway within lysosomes (Sherman and Goldberg 2001).
Inducible HSPs prevent the emergence of protein aggregation events during cell exposure to many stress stimuli, including a variety of central nervous system insults, such as cerebral ischemia, seizures, neurotoxin exposure, and other physiochemical insults (Muchowski and Wacker 2005).
A recent study in the US population showed that the overall level of constitutively expressed HSPs in different classes of neurons, seems intimately related to the age of onset, kinetics of progression, and severity of disease, since it correlates with the relative frequencies of AD, PD, and ALS (Chen and Brown 2007). Indeed, recent observations in the in vitro and in vivo models of AD, PD and HD, indicate that HSPs are potent suppressors of cell death evoked by toxic aggregates of disease-specific proteins, such as tau, beta-amyloid1-42, alpha-synuclein and huntingtin (Kopito and Ron 2000).
Based on this, novel therapeutic approaches have been suggested, aimed to potentiate cell stress response by enhancing the activation of HSPs against misfolded or aggregation-prone proteins (Chaudhuri and Paul 2006).
Interestingly, small HSPs, namely, Hsp20 and Hsp27, have been found in pathological protein aggregates, that are a typical feature of several neurodegenerative disorders (Renkawek et al. 1994; Wilhelmus et al. 2006). Furthermore, Hsp20, as well as other small HSPs, such as alphaB-crystallin, Hsp27, and HspB2, have been shown to act as in vitro lysine- and/or glutamine-donors for TG2, and to be readily cross-linked with beta-amyloid1-40 (Boros et al. 2004). More recently, Hsp20 has been shown to be easily transamidated and deamidated by TG2 in the in vitro experiments (Boros et al. 2006). Some in vivo observations have also demonstrated the cross-linking of (poly)ubiquitin moieties to parkin and alpha-synuclein, via Hsp27, by TG-catalysed γ-glutamyl-ε-lysine bonds in neurofibrillary tangles of AD brains (Nemes et al. 2004).
In the light of these findings, some experimental work was carried out in our lab to test the hypothesis of an interaction between TG2 and small HSPs in cell response to neurodegeneration.
Preliminary results (unpublished) show that excitotoxic doses of N-methyl-d-aspartate (NMDA) caused a strong reduction of basal Hsp20 and Hsp27 levels in retinoic acid (RA)-differentiated human neuroblastoma cells, SH-SY5Y, expressing high levels of TG2. Notably, NMDA effects were reversed in the presence of R283, a site-specific inhibitor of TG activity (Griffin et al. 2004). Immunoprecipitation experiments demonstrated an interaction between TG2 and Hsp27, and the occurrence of Hsp27 macromolecular assemblies that was partially prevented by TG activity inhibition.
These observations seem to suggest a likely involvement of TG activity in the stabilization of intra-neuronal protein aggregates by interference with the proteasome-mediated degradation of unfolded proteins. Further investigations are needed to elucidate this topic.
Conclusions
In the last years, several reports have significantly contributed to our knowledge of TG2 role in the mechanisms of neurodegeneration. However, to date it has not yet been clarified whether TG2 plays a protective or adverse role in neuronal cell death.
Further studies aimed to the identification of TG substrates, and a deeper characterization of the relationships between TG2 and other stress proteins, such as HSPs, would be very helpful in the perspective to improve the therapeutic management of neurodegenerative disorders.
Therapeutic strategies to decrease neurotoxicity have so far included the use of antibodies, caspase inhibitors, and various chemical inhibitors.
Since proteolytic stress plays a pivotal role in the onset of neuronal damage, it has recently been proposed that neurodegenerative disorders could be cured by preventing protein misfolding and aggregation, or by returning misfolded proteins to normal, or by accelerating the degradation of aggregated proteins.
In this perspective, the use of transglutaminase inhibitors or the stimulation of the HSP activation pathway, preventing aggregation processes, may result in beneficial effects against neurodegeneration.
Abbreviations
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- CSF:
-
Cerebrospinal fluid
- ECM:
-
Extracellular matrix
- GGEL:
-
γ-Glutamyl-ε-lysine
- HD:
-
Huntington’s chorea
- HSPs:
-
Heat shock proteins
- NF-kappa B:
-
Nuclear factor-kappa B
- NMDA:
-
N-methyl-d-aspartate
- RA:
-
Retinoic acid
- SCA:
-
Spinocerebellar ataxia
- SBMA:
-
Spinobulbar muscular atrophy
- TG(s):
-
Transglutaminase(s)
- TG2:
-
Tissue transglutaminase
References
Andringa G, Lam KY, Chegary M, Wang X, Chase TN, Bennett MC (2004) Tissue transglutaminase catalyzes the formation of alpha-synuclein crosslinks in Parkinson’s disease. FASEB J 18:932–934
Bailey CD, Johnson GV (2005) Tissue transglutaminase contributes to disease progression in the R6/2 Huntington’s disease mouse model via aggregate-independent mechanisms. J Neurochem 92:83–92
Battaglia G, Farrace MG, Mastroberardino PG et al (2007) Transglutaminase 2 ablation leads to defective function of mitochondrial respiratory complex I affecting neuronal vulnerability in experimental models of extrapyramidal disorders. J Neurochem 100:36–49
Bonelli RM, Aschoff A, Niederwieser G, Heuberger C, Jirikowski G (2002) Cerebrospinal fluid tissue transglutaminase as a biochemical marker for Alzheimer’s disease. Neurobiol Dis 11:106–110
Boros S, Kamps B, Wunderink L, de Bruijn W, de Jong WW, Boelens WC (2004) Transglutaminase catalyzes differential crosslinking of small heat shock proteins and amyloid-beta. FEBS Lett 576:57–62
Boros S, Ahrman E, Wunderink L, Kamps B, de Jong WW, Boelens WC, Emanuelsson CS (2006) Site-specific transamidation and deamidation of the small heat-shock protein Hsp20 by tissue transglutaminase. Proteins 62:1044–1052
Caccamo D, Currò M, Cusumano G, Crisafulli G, Ientile R (2004) Excitotoxin-induced changes in transglutaminase during differentiation of cerebellar granule cells. Amino Acids 26:197–201
Caccamo D, Campisi A, Currò M, Aguennouz M, Li Volti G, Avola R, Ientile R (2005) Nuclear factor-kappaB activation is associated with glutamate-evoked tissue transglutaminase up-regulation in primary astrocyte cultures. J Neurosci Res 82:858–865
Campisi A, Caccamo D, Raciti G, Cannavò G, Macaione V, Currò M, Macaione S, Vanella A, Ientile R (2003) Glutamate-induced increases in transglutaminase activity in primary cultures of astroglial cells. Brain Res 978:24–30
Campisi A, Caccamo D, Li Volti G, Currò M, Parisi G, Avola R, Vanella A, Ientile R (2004) Glutamate-evoked redox state alterations are involved in tissue transglutaminase upregulation in primary astrocyte cultures. FEBS Lett 578:80–84
Chaudhuri TK, Paul S (2006) Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J 273:1331–1349
Chen S, Brown IR (2007) Neuronal expression of constitutive heat shock proteins: implications for neurodegenerative diseases. Cell Stress Chaperones 12:51–58
Currò M, Condello S, Caccamo D, Ferlazzo N, Parisi G, Ientile R (2009) Homocysteine-induced toxicity increases TG2 expression in Neuro2a cells. Amino Acids 36:725–730
Dudek SM, Johnson GV (1994) Transglutaminase facilitates the formation of polymers of the beta-amyloid peptide. Brain Res 651:129–133
Griffin M, Casadio R, Bergamini CM (2002) Transglutaminases: nature’s biological glues. Biochem J 368:377–396
Griffin M, Coutts IG, Saint R (2004) International Publication Number WO 2004/1133603, GB patent PCT/GB2004/002569
Ientile R, Caccamo D, Macaione V, Torre V, Macaione S (2002) NMDA-evoked excitotoxicity increases tissue transglutaminase in cerebellar granule cells. Neuroscience 115:723–729
Ientile R, Caccamo D, Marciano MC, Currò M, Mannucci C, Campisi A, Calapai G (2004) Transglutaminase activity and transglutaminase mRNA transcripts in gerbil brain ischemia. Neurosci Lett 363:173–177
Ientile R, Caccamo D, Griffin M (2007) Tissue transglutaminase and the stress response. Amino Acids 33:385–394
Jeitner TM, Bogdanov MB, Matson WR et al (2001) N(epsilon)-(gamma-l-glutamyl)-l-lysine (GGEL) is increased in cerebrospinal fluid of patients with Huntington’s disease. J Neurochem 79:1109–1112
Jeitner TM, Matson WR, Folk JE, Blass JP, Cooper AJ (2008) Increased levels of gamma-glutamylamines in Huntington disease CSF. J Neurochem 106:37–44
Jeitner TM, Pinto JT, Krasnikov BF, Horswill M, Cooper AJL (2009) Transglutaminases and neurodegeneration. J Neurochem 109:160–166
Johnson GV, LeShoure R Jr (2004) Immunoblot analysis reveals that isopeptide antibodies do not specifically recognize the epsilon-(gamma-glutamyl)lysine bonds formed by transglutaminase activity. J Neurosci Methods 134:151–158
Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM (2003) Tissue transglutaminase-induced aggregation of alpha-synuclein: implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 100:2047–2052
Kim SY, Grant P, Lee JH, Pant HC, Steinert PM (1999) Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer’s disease. J Biol Chem 274:30715–30721
Kopito RR, Ron D (2000) Conformational disease. Nat Cell Biol 2:E207–E209
Lee J, Kim YS, Choi DH, Bang MS, Han TR, Joh TH, Kim SY (2004) Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J Biol Chem 279:53725–53735
Matthew A, Morimoto RI (1998) Role of the heat-shock response in the life and death of proteins. Ann N Y Acad Sci 851:99–111
Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nature Rev Mol Cell Biol 1:120–129
Mattson MP, Camandola S (2001) NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 107:247–254
Miller ML, Johnson GV (1995) Transglutaminase cross-linking of the tau protein. J Neurochem 65:1760–1770
Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22
Muma NA (2007) Transglutaminase is linked to neurodegenerative diseases. J Neuropathol Exp Neurol 66:258–263
Nemes Z, Devreese B, Steinert PM, VanBeeumen J, Fesus L (2004) Cross-linking of ubiquitin, HSP27, parkin, and alpha-synuclein by gamma-glutamyl-epsilon-lysine bonds in Alzheimer’s neurofibrillary tangles. FASEB J 18:1135–1137
Renkawek K, Bosman GJ, de Jong WW (1994) Expression of small heat-shock protein hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol 87:511–519
Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10(Suppl):S10–S17
Segers-Nolten IM, Wilhelmus MM, Veldhuis G, van Rooijen BD, Drukarch B, Subramaniam V (2008) Tissue transglutaminase modulates alpha-synuclein oligomerization. Protein Sci 17:1395–1402
Sherman MY, Goldberg AL (2001) Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29:15–32
Siegel M, Khosla C (2007) Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol Ther 115:232–245
Singer SM, Zainelli GM, Norlund MA, Lee JM, Muma NA (2002) Transglutaminase bonds in neurofibrillary tangles and paired helical filament tau early in Alzheimer’s disease. Neurochem Int 40:17–30
Soto C, Estrada LD (2008) Protein misfolding and neurodegeneration. Arch Neurol 65:184–189
Tucholski J, Kuret J, Johnson GV (1999) Tau is modified by tissue transglutaminase in situ: possible functional and metabolic effects of polyamination. J Neurochem 73:1871–1880
Van Raamsdonk JM, Pearson J, Bailey CD, Rogers DA, Johnson GV, Hayden MR, Leavitt BR (2005) Cystamine treatment is neuroprotective in the YAC128 mouse model of Huntington disease. J Neurochem 95:210–220
Vermes I, Steur EN, Jirikowski GF, Haanen C (2004) Elevated concentration of cerebrospinal fluid tissue transglutaminase in Parkinson’s disease indicating apoptosis. Mov Disord 19:1252–1254
Wilhelmus MM, Otte-Holler I, Wesseling P, de Waal RM, Boelens WC, Verbeek MM (2006) Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol Appl Neurobiol 32:119–130
Zainelli GM, Ross CA, Troncoso JC, Muma NA (2003) Transglutaminase cross-links in intranuclear inclusions in Huntington disease. J Neuropathol Exp Neurol 62:14–24
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Caccamo, D., Currò, M., Condello, S. et al. Critical role of transglutaminase and other stress proteins during neurodegenerative processes. Amino Acids 38, 653–658 (2010). https://doi.org/10.1007/s00726-009-0428-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-009-0428-3