



Abstract
Transplantation of neural stem cells (NSCs) is a novel strategy to restore function in the diseased brain, acting through multiple mechanisms, for example, neuronal replacement, neuroprotection, and modulation of inflammation. Whether transplanted NSCs can operate by fusing with microglial cells or mature neurons is largely unknown. Here, we have studied the interaction of a mouse embryonic stem cell-derived neural stem (NS) cell line with rat and mouse microglia and neurons in vitro and in vivo. We show that NS cells spontaneously fuse with cocultured cortical neurons, and that this process requires the presence of microglia. Our in vitro data indicate that the NS cells can first fuse with microglia and then with neurons. The fused NS/microglial cells express markers and retain genetic and functional characteristics of both parental cell types, being able to respond to microglia-specific stimuli (LPS and IL-4/IL-13) and to differentiate to neurons and astrocytes. The NS cells fuse with microglia, at least partly, through interaction between phosphatidylserine exposed on the surface of NS cells and CD36 receptor on microglia. Transplantation of NS cells into rodent cortex results in fusion with mature pyramidal neurons, which often carry two nuclei, a process probably mediated by microglia. The fusogenic role of microglia could be even more important after NSC transplantation into brains affected by neurodegenerative diseases associated with microglia activation. It remains to be elucidated how the occurrence of the fused cells will influence the functional outcome after NSC transplantation in the diseased brain.
Introduction
Transplantation of stem cells holds much promise as potential novel treatment to restore function in human brain disorders. Studies in animal models have indicated that transplantation of neural stem cells (NSCs) improves function by multiple mechanisms such as replacing lost neurons, trophic actions, modulation of inflammation, stimulation of angiogenesis, and neuroprotection [1]. Clonogenic neural stem (NS) cell lines derived from embryonic stem (ES) cells or rodent and human fetal brain retain multilineage differentiation capacity after prolonged expansion [2] and give a high yield of neurons [3]. These features make NS cells a very attractive source for intracerebral transplantation.
Neurodegenerative diseases are associated with inflammation, that is, activation of innate immune cells, such as microglia, and infiltration of monocytes and other immune cells from the peripheral blood. Microglia/macrophages probably exert a dual action in the injured or diseased brain, either by exacerbating the damage or by promoting repair [4]. Microglia, like other cells of the monocytic lineage [5] can fuse to each other [6] and with mature resident neurons [7]. Bone marrow-derived cells (BMDCs) transplanted intraperitoneally in rodents [8] or intravenously in humans [9] fuse with cerebellar Purkinje neurons. Fusion might have a protective role [8] as it is markedly enhanced by inflammation and tissue damage [10–12]. Several examples of fusion between stem/progenitor cells have also been reported. ES cells can fuse with fetal and adult mouse brain-derived progenitors in vitro, the tetraploid hybrids being fully pluripotent, including multilineage contribution to chimeras [13]. NSCs from neonatal mouse subventricular zone (SVZ), adult mouse whole brain, or adult rat hippocampus have been shown to fuse with each other, but the fused cells do not divide and cannot be propagated [14, 15].
Little is currently known about the potential for fusion between NSCs and microglial cells or mature neurons in vivo after transplantation. Such knowledge is highly warranted for understanding the mode of action of grafted NSCs. Here, we studied the interaction between mouse ES cell-derived NS cells and mouse and rat neurons or microglia. We demonstrate that these cells spontaneously fuse in vitro, giving rise to cell hybrids with characteristics of their parental cells. We also found that microglia mediate fusion of NS cells with mature neurons in vitro showing a new role for this cell type. Recapitulating what we observed in culture, NS cells fuse with mature neurons also in vivo after transplantation into neonatal rat and mouse cortex.
Materials and Methods
Derivation and handling of human fetal tissue and all experimental procedures were approved by the local Ethics Committees and were conducted in accordance with European Union directives.
Cell Cultures
Mouse NS Cell Culture
Mouse ES cell-derived NS cells (LC1) were maintained in NS medium (NS-med) (containing Euromed [Euroclone, Milano, Italy, http://www.euroclonegroup. it], N2 supplement [Gibco, Grand Island, NY, http://www.invitrogen.com], 20 ng/ml epidermal growth factor (EGF) [R&D Systems, Abingdon, UK, http://www.rndsystems.com] and 20ng/ml basic fibroblast growth factor (bFGF) [R&D Systems]) on uncoated plastic [2]. Mouse fetal cortex-derived NS cells (Cor1) were grown on laminin (Invitrogen, Carlsbad, CA, http://www.invitrogen.com)-coated flasks (3 μg/ml) in the same medium. Unless otherwise stated, we will refer to ES-derived LC1 cells as NS cells. For experiments, both cell lines were infected with CAG-GFP or CAG-RFP retrovirus [16]. For transplantation, cells were resuspended at 100,000 cells per μl in Hanks Balanced Saline Solution (Gibco).
Mouse and Rat Microglia Culture
Primary microglia were isolated as previously described [17] from brains of neonatal Wistar rats (Taconic, Bomholt, Denmark, http://www.taconic.com) or transgenic mice expressing green fluorescent protein (GFP) under chicken β-actin promoter [18]. Primary cultures were maintained in microglia medium (Micro-med) consisting of Dulbecco's modified Eagle's medium (DMEM)/F12 supplemented with Glutamax, 10% fetal bovine serum (FBS), and penicillin-streptomycin (all from Gibco). Microglia cells were plated on poly-L-lysine (PLL)-coated chamber-slides (Lab-Tek, Nunc A/S, Denmark, http://www.nuncbrand.com) or glass coverslips (20,000 cells per cm2). These cultures contained 93.1% ± 1.4% Iba1+ microglia, and 1.0% ± 0.8% glial fibrillary acidic protein (GFAP)+ astrocytes. In case of rat microglia, cells were labeled using PKH26 (Sigma, Stockholm, Sweden, http://www.sigma-aldrich.com) according to manufacturer's instructions. BV2 mouse microglia cell line was obtained from Interlab Cell Line Collection (Genova, Italy). Cells were grown on uncoated plastic flasks in a medium based on RMPI 1640 (Gibco), containing 10% FBS (Gibco) and 2 mM glutamine (Gibco). The BV2 cells could be maintained in NS-med, without any noticeable sign of cell death or damage. For experiments, BV2 cells were infected with either CAG-GFP or CAG-RFP retrovirus.
Mouse and Rat Primary Cortical Cell Culture
Cortical tissue from E17 β-actin-GFP mice [18] or E18 Wistar rats (Taconic) was used to isolate primary cells. These cells were plated on poly-D-lysine (PDL)/laminin-coated slides at 20,000 cells per cm2 in primary medium (Pri-med) containing Neurobasal (Gibco), 2% B27 (Gibco), 0.5 mM glutamine (Gibco), and penicillin-streptomycin (Sigma).
Human Cultures
Forebrain tissue and cortical neurospheres from dead aborted human fetuses 8 weeks postconception were derived as previously reported [19]. Primary cortical human fetal-derived cultures were plated on PDL/laminin-coated glass coverslips (20,000 cells per cm2) and maintained in Pri-med without antibiotics. Human cortex-derived NS culture [20] was maintained in NS-med and plated onto laminin-coated flasks. For some experiments human cortical NS were transduced with CAG-GFP retrovirus. Human induced pluripotent stem (iPS) cell-derived neuroepithelial-like stem cells (lt-NES) cells were expanded and cultured as previously described [21].
Cultures of Fused Cells
Sorted GFP+/RFP+ cells were cultured in NS-med, plated onto poly-L-ornithine/laminin-coated flasks, and passaged every 2–3 days using Accutase (PAA, Linz, Austria, http://www.paa.at). The LC1 multistep differentiation protocol previously described [3] was applied with minor modifications to induce neuronal differentiation of the fused cells. Sorted fused CellVue+/GFP+/RFP+ cells were replated onto PDL/laminin-coated glass coverslips and maintained for 7 days in Pri-med.
Cocultures
Primary Cells and NS Cell Coculture
Seven to twelve days after plating primary cells, NS cells were plated on top (10,000 cells per cm2) in Pri-med or NS-med for 1–3 days. For one set of experiments, rat primary cells were treated with 10 nM Mac1-Saporin (Advanced Targeting Systems, San Diego, CA, http://www.atsbio.com/) or control mouse anti-Mac1 antibody (Advanced Targeting Systems) during the 5 days prior to the coculture, and analyzed 3 days thereafter.
Primary cortical cells, isolated from E17 Wistar rats, were labeled with PKH-26 (Sigma) and plated on PDL/laminin-coated glass coverslips (20,000 cells per cm2) in Pri-med. After 12 days of culture, cells were treated with 10 nM Mac1-Saporin for 4 days. Primary microglia isolated from P2 Wistar rats were plated on half of the microglia-depleted primary cortical cultures at 20,000 cells per cm2 and cocultured for 5 days. A fraction of isolated microglia analyzed before the coculture showed that virtually all cells were immunopositive for the microglia marker Iba1. After 5 days of coculturing, GFP+ NS cells were plated onto the microglia-depleted primary cultures for additional 4 days.
Microglia and NS Cell Coculture
NS cells were plated on microglial cultures as described above, maintained in NS-med, and analyzed 1–5 days thereafter. To label microglia with bromo-2-deoxyuridine (BrdU), cells were cultured for 5 days with 10 μM BrdU (BD Biosciences, San Diego, CA, http://www.bdbiosciences.com). For AnnexinV experiments, cocultures were performed in presence of 2.5% AnnexinV (Invitrogen).
Primary Cell and Microglia Coculture
GFP+ microglia were plated on 7–12 days old primary cell cultures at a density of 20,000 cells per cm2 in Pri-med and analyzed 1–3 days thereafter.
Mouse Fetal Cortex-Derived Neurospheres and Microglia Treated with Interleukin (IL)-4 or Vehicle
Mouse fetal cortex-derived neurospheres [2] infected with CAG-retrovirus were plated onto PLL-coated flasks at about 20,000 cells per cm2. After 24 hours, RFP+ microglia were plated on the top of attached neurospheres (20,000 cells per cm2). Cocultures were maintained under adherent proliferation conditions in NS-med 3–6 days and treated daily with IL-4 10 ng/ml (R&D Systems, Minneapolis, MN, http://www.rndsystems.com). After 3–5 days, cocultures were disaggregated to single cells and plated onto poly-D-ornithine/laminin-coated glass coverslips (20,000 cells per cm2). After 1 hour, cells were fixed and processed for immunocytochemistry.
Mouse-Derived Cortical Cells and Fused NS/Microglia Cells
Primary cortical cultures were derived from postnatal day 1 Balb/c mouse. Cells were labeled with CellVue Claret (Sigma) and plated onto PDL/laminin-coated glass coverslips (20,000 cells per cm2) and maintained in Pri-med. After 20 days, fused NS/microglia (GFP+/RFP+) cells (20,000 cells per cm2) were plated on top of the primary cortical cultures in Pri-med and cultured for 5 days, and then processed for fluorescent-activated cell sorting (FACS).
Human-Derived Cells and Microglia
Human fetal cortex-derived GFP+ cells expanded as neurospheres, human fetal cortex-derived cells expanded as NS cells, and human iPS cell-derived lt-NES cells were plated on PDL, PDL laminin-, or PLL-coated flasks, respectively, at a density of approximately 20,000 cells per cm2. After 24 hours, RFP+ microglia cells were plated on top of the cultures (20,000 cells per cm2). After 3–5 days, cocultures were analyzed for fusion using immunocytochemistry.
Human-Derived Cells and Fused NS/Microglia Cells
Differentiated human iPS cell-derived lt-NES cells labeled with CellVue and human fetal cortex-derived primary cortical cells were plated (20,000 cells per cm2) onto poly-L-ornithine/laminin- and PDL laminin-coated glass coverslips, respectively. After 20 days of culture, GFP+/RFP+ fused NS/microglia cells were plated on top of the primary cultures, cocultured for 4–5 days, and then analyzed thereafter by immunocytochemistry.
Animal Procedures
Transplantation in Rat and Mouse Pups
Two to 3 days old Wistar rat pups (n = 8) were injected with NS cell suspension (1 μl per deposit) unilaterally into cortex [19] (coordinates: AP +0.5; L −2.0; V −0.8 mm from bregma) and sacrificed 1 (n = 4) or 4 weeks (n = 4) thereafter. Four to 5 days old C57/black6 mouse pups (Taconic) were subjected to a similar transplantation procedure (coordinates: AP +2.5; L −1.1; V −0.6 mm from lambda) and processed after 1 (n = 3), 2 (n = 3), 4 (n = 8), 6 (n = 6), or 8 (n = 4) weeks.
Lumafluor Injection
Two days old C57/black6 mouse pups (n = 4) were injected with 0.4 μl of Lumafluor RedBeads (Lumafluor, Naples, FL, https://lumafluor.com) in the left dorsolateral striatum (coordinates AP +2.1; L +1.2; V −1.2). Two days later, animals received transplants in the right cortex as described above and were sacrificed 8 weeks thereafter.
Fluorescence-Activated Cell Sorting
FACS was performed on FACSAria SORP system with FACS Diva v6.0 software (BD Biosciences). Doublets were excluded via gating through both forward scatter-height versus width and side scatter-height versus width. The gate identifying fused (GFP+/RFP+) cells was set according to non-GFP/RFP expressing controls. Gate locations were verified using cocultures of non-GFP/RFP expressing cells. The gate identifying triple GFP+/RFP/+CellVue+ cells was set according to fused GFP+/RFP+ cultures and CellVue-labeled mouse cortical cells. Gate locations were verified using cultures of fused GFP+/RPF+/CellVue+ cells as well as cocultures of microglia (RFP+), NS (GFP+), and cortical (CellVue+) cells and unlabeled cocultures of microglia, NS, and cortical cells. Correct sorting of the gated double- and triple-positive cell population was confirmed by FACS reanalysis and fluorescence microscopy. Flow cytometric data were analyzed using FlowJo software (Tree star, Ashland, OR, http://www.flowjo.com).
Immunocytochemistry
Cell cultures were fixed using 4% paraformaldehyde (PFA) for 20 minutes, with addition of 0.2% glutaraldehyde for GABA staining. Animals were transcardially perfused with 4% PFA, and the brains were cryoprotected and cut on a freezing microtome into 30-μm-thick coronal sections. For both cells and sections, nonspecific binding was blocked using a 5% solution of the appropriate serum in buffer, containing 0.025% or 0.25% Triton, respectively. Incubation with primary antibodies was performed overnight at 4°C. The following antibodies were used: mouse, rabbit, and chicken anti-GFP (1:10,000 Abcam [Cambridge, U.K., http://www.abcam.com], 1:10,000 Abcam, and 1:3,000 Millipore [Billerica, MA, http://www.millipore.com], respectively), biotinylated or unconjugated mouse anti-NeuN (1:200 Millipore), rabbit and mouse anti-RFP (1:1,000 Abcam and 1:500 Abcam, respectively), rat anti-M2/M6 (1:50 Hybridoma Bank, Iowa, IA, http://www.uiowa.edu/∼dshbwwwD), rabbit and goat anti-Iba1 (1:1000 Wako [Osaka, Japan, http://www.wako-chem.co.jp/english] and 1:200 Millipore, respectively), rat anti-BrdU (1:100 AbD Serotec), mouse anti-rat Mac1 (1:100 AbD Serotec, Oxford, UK, http://www.abdserotec.com), mouse anti-MAP2 (1:500 Sigma), rabbit anti-v-Glut-1 (1:500 Synaptic Systems, Goettingen, Germany, http://www.sysy.com), mouse anti-GABA (1:200 Sigma), mouse and rabbit anti-GFAP (1:500 Sigma and1:400 Dako [Glostrup, Denmark, http://www.dako.com], respectively), mouse anti-MBP (1:200, Millipore), mouse anti rat CD68 (ED1) (1:200 AbD Serotec), mouse anti human cytoplasm SC121 (1:500 provided by Dr N. Uchida, StemCells, Inc., Palo Alto, CA, USA), and rabbit anti-Olig2 (1:300, Abcam). Primary antibodies were detected with appropriate fluorescent or biotinylated secondary antibodies. In the latter case, the reaction was eventually developed with the appropriate fluorophore-conjugated streptavidin (Cy3, Cy5 [Jackson Immunolabs, West Grove, PA, http://www.jacksonimmuno.com] or Alexa488 [Invitrogen]). Hoechst 33342 (1:1,000, Invitrogen) or TO-PRO-3 (1:1,000, Invitrogen) were used to label cell nuclei.
For AnnexinV staining, cells were incubated with biotin-conjugated AnnexinV (1:50,) in binding buffer (10 mM HEPES, pH 7.5, containing 140 mM NaCl and 2.5 mM CaCl2) for 10 minutes. After washing, fluorescent streptavidin was used to detect AnnexinV. As negative control, the procedure was performed without Ca2+, which is required for binding of AnnexinV.
Microscopy and Quantitative Analysis
Specimens were analyzed using either fluorescence microscopy (Olympus, Ballerup, Denmark, http://www.olympus.dk) or laser scanning confocal microscopy (Leica, Wetzlar, Germany, http://www.leica.com/). Imaging of living cells was performed in an inverted microscope (Zeiss, Oberkochen, Germany, http://www.zeiss.com). Cell counting in in vitro experiments was carried out by a blinded observer using stereology with randomized sampling system (C.A.S.T.-GRID, Olympus). At least 200 cells per slide were counted for general characterizations and at least 500 cells were analyzed when quantifying fusion. In Mac1-Saporin experiments, more than 1,500 cells were counted. Two-tailed unpaired t test and one-way ANOVA, followed by Dunnett post hoc test were used to assess differences between the groups. Data are expressed as means ± SEM.
Electrophysiology
Recordings were performed as previously described [22]. Briefly, cells plated on coverslips were constantly perfused with heated (32–34°C), gassed (95% O2, 5% CO2) solution (pH 7.2–7.4, 295–300 mOsm) containing (in mM): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 26 NaHCO3, 1.25 NaH2PO4, and 25 glucose. Recording pipettes were filled with solution (pH 7.2–7.4, 295–300 mOsm) containing (in mM): 122.5 potassium gluconate, 12.5 KCl, 10.0 KOH-HEPES, 0.2 KOH-EGTA, 2 MgATP, 0.3 Na3-GTP, and 8 NaCl, resulting in pipette resistances of 3–5 MΩ. Biocytin (0.5%; Sigma) was freshly dissolved in the pipette solution before recordings for post hoc identification of recorded cells.
Results
Microglia Contribute to Fusion of NS Cells with Primary Cortical Neurons In Vitro
To explore whether mouse ES cell-derived NS cells can fuse with neurons, we first cultured primary cortical cells from β-actin-GFP mouse embryos. After 7–12 days, all cells were GFP+, 79.2% ± 3.7% were MAP2+ neurons, 10.8% ± 2.3% were GFAP+ astrocytes, and 5.6% ± 1.9% were Iba1+ microglia. Virtually no oligodendrocytes were found. We then plated RFP+ NS cells onto these cultures. After 3 days of coculture, 5.2% ± 1.2% of cells expressed both GFP and RFP, detected both with specific antibodies and by endogenous fluorescence. The majority of double-labeled cells also expressed neuron-specific markers, such as NeuN (Fig. 1A–1D), v-Glut1, or MAP2 (data not shown) and had the appearance of differentiated mature neurons. This was observed already after 3 days of coculturing, and never when only NS cells were cultured for the same duration, strongly indicating that the NS cells had fused with the primary cortical neurons. Moreover, 27.9% ± 6.1% of the GFP+/RFP+ cells had two nuclei, further suggesting that they had fused.
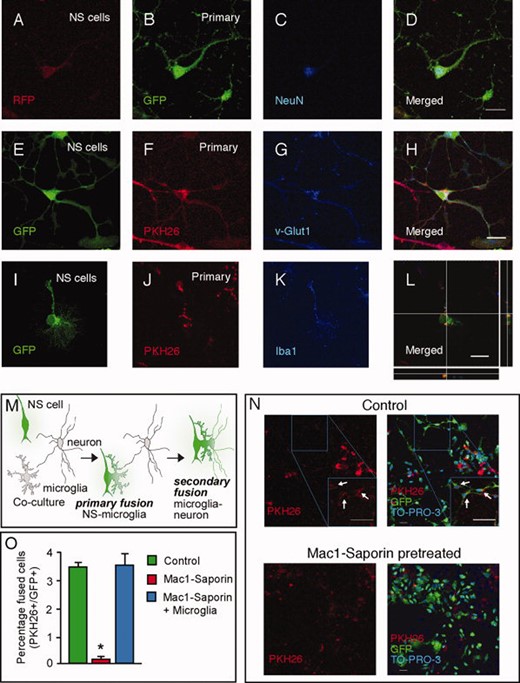
Embryonic stem (ES)-derived NS cells fuse with neurons in primary cortical cultures only in presence of microglia. (A–L): Mouse or rat primary cortical cells cocultured with NS cells fuse and express neuronal (NeuN and v-Glut1) or microglial markers (Iba1). (M): Hypothetical model of the fusion process in the cocultures. (N,O): Removal of microglia by pretreatment with Mac1-Saporin results in almost complete absence of fused double-labeled cells (arrows) in the cocultures, and reintroduction of microglia fully restored fusion rate. Data are given as means ± SEM. *, p < .05, unpaired t test. Scale bars: D, H, and L = 20 μm, n = 50 μm. Abbreviations: NS, neural stem; GFP, green fluorescent protein; RFP, red fluorescent protein.
Similar results were obtained when coculturing PKH26-labeled rat primary cortical cells with GFP+ NS cells (Fig. 1E–1H). Rat primary cortical cultures had a cellular composition similar to that of mouse cultures, and we found that several of the double-positive cells (28.3% ± 4.4%) were carrying two nuclei. Thus, the formation of double-positive cells was not dependent on the species of cell origin or the cell-labeling method. We detected a few RFP+/GFP+ or GFP+/PKH26+ cells which were also positive for the microglial marker Iba1 (Fig. 1I–1L), but none of the cells stained for the astrocyte marker GFAP (data not shown).
The primary cortical cultures contained microglia, and microglia/macrophages are known for their fusion capacity [23, 24] also with cortical neurons in vivo [7]. We speculated that microglia could be involved in the fusion between NS cells and primary cortical neurons in vitro (Fig. 1M). To test this hypothesis, we used the microglia-specific toxin Mac1-saporin to ablate microglia from PKH26-labeled rat primary cortical cultures, which were then cocultured with GFP+ NS cells. Pretreatment with Mac1-saporin killed virtually all Iba1+ microglia (data not shown). Three days after plating NS cells on primary cortical cell cultures lacking microglia, the number of fused GFP+/PKH26+ cells was reduced from 3.5% ± 0.1% in cultures containing microglia to 0.1% ± 0.1% in microglia-depleted cultures (Fig. 1N, 1O). Reintroduction of microglia restored the decreased level of fusion caused by microglia ablation back to the initial level (3.6% ± 0.4%) (Fig. 10).
To explore whether NS cells fuse with microglia, we cocultured GFP+ NS cells with RFP+ microglia (BV2 cell line). In support, a large portion (26.8% ± 4.1%) of the cells became positive for both GFP and RFP after 2 days of coculturing. Fusion between NS cells and microglia (Fig. 2A–2D) was confirmed by several other experiments. First, when coculturing primary mouse (GFP+) or rat (PKH26-labeled) microglia with RFP+ or GFP+ NS cells, respectively, about one third of the cells expressed both markers and, of these, 31.8% ± 1.8% contained two nuclei (Fig. 2E–2H). In rat microglia cultures, the double-labeled cells were stained with the mouse-specific antibodies M2/M6 (Fig. 2I–2L), indicating that the fused cells retained some characteristics of NS cells. Second, we provided evidence against nonspecific transfer of fluorophores from microglia to NS cells by prelabeling proliferating GFP+ microglia with BrdU. After 5 days of coculturing of GFP+ microglia with RFP+ NS cells, we detected a substantial portion of cells triple-positive for GFP, RFP, and BrdU (Fig. 2M–2P). Third, to rule out that the acquisition of RFP in GFP+ microglia was due to phagocytosis, we cocultured microglia with NS cells which had been killed by freezing–thawing cycles. Arguing against phagocytosis, we found no GFP+/RFP+ cells in these cocultures (not shown). Fourth, we demonstrated healthy-looking GFP+/RFP+ cells in living microglia-NS cell co-cultures (Fig. 2Q–2S). Fifth, we showed that the percentage of GFP+/RFP+ cells increased over time after 1, 2, 3, and 5 days of coculturing GFP+ microglia and RFP+ NS cells (Fig. 2T). This observation suggests that fusion between microglia and NS cells in vitro is a spontaneously and continuously occurring process. Sixth, we performed similar cocultures of either BV2 or primary mouse microglia (RFP+ and GFP+, respectively) with mouse fetal cortex-derived NS cells (Cor1), labeled with GFP or RFP. Also in this case, we readily identified fused cells expressing both markers (Fig. 2U–2Z3), although at a lower rate as compared to ES cell-derived NS cells. These data indicate that fusion with microglia is not restricted to ES-derived NS cells but also occurs with fetal cortex-derived NS cells.
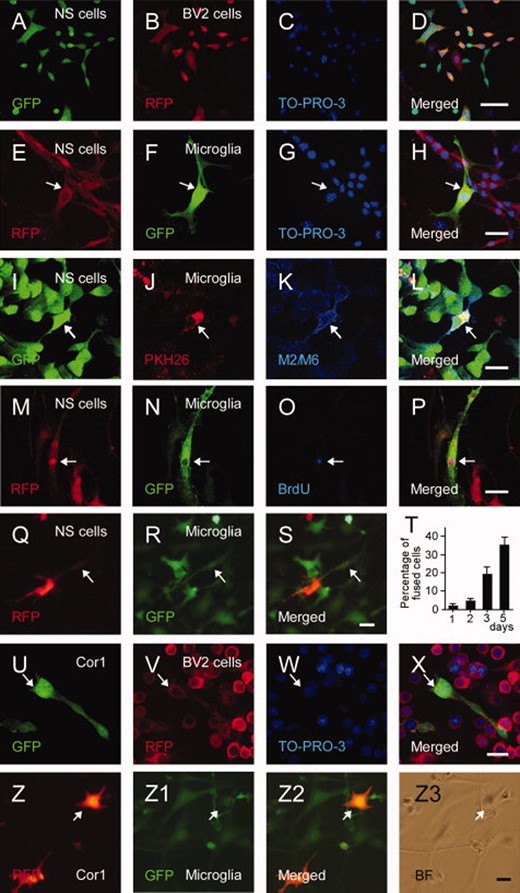
Embryonic stem (ES)-derived NS cells fuse in vitro with microglia of different sources. (A–D): Coculturing RFP+ BV2 cell line and GFP+ NS cells leads to fusion as observed in confocal images. Similar results were obtained with primary mouse (E–H) or rat (I–L) microglia. (I–L): Mouse ES-derived GFP+ NS cells fused with rat PKH26+ microglia maintain expression of the mouse-specific markers M2/M6. (M–P): Fused GFP- and BrdU-labeled microglia and RFP+ NS cells are triple-positive for GFP, BrdU, and RFP. (Q–S): Living cell images of cocultures of GFP+ microglia and RFP+ NS cells. (T): Percentage of fused cells in the cocultures of GFP+ microglia and RFP+ NS cells. Means ± SEM. (U–Z3): Fusion of Cor1 fetal cortex-derived NS cells (Cor1) with BV2 cells (U–X) or primary mouse microglia (Z–Z3). Scale bars = 50 μm (D), 20 μm (H, L, P, S, X and Z3). Abbreviations: BrdU, bromo-2-deoxyuridine; GFP, green fluorescent protein; NS, neural stem; RFP, red fluorescent protein.
Fused NS/Microglia Cells Display Morphological Properties of NS Cells
To be able to analyze in more detail the characteristics of the fused cells, we used FACS to separate GFP+/RFP+ cells from single-positive GFP+ NS cells and RFP+ microglia fractions (Fig. 3A). The three populations of cells were easily identified (Fig. 3A). We assessed the efficiency of cell sorting by analyzing the fluorescence of the cells after 6 hours of reculturing following sorting. The fractions with GFP+/RFP+ cells (Fig. 3C) and RFP+ microglia (Fig. 3D) were virtually pure. Almost all cells in the GFP+ fraction (Fig. 3E) were NS cells but rare GFP+/RFP+ cells were also detected, probably because of the low sensitivity of the FACS equipment we used to detect RFP fluorescence. After initial replating, the sorted fused cells as well as the NS cells and microglia continued to proliferate (Supporting Information Fig. S1), maintaining their respective fluorescence profile for at least 1 week (Fig. 3F). The fused cells could be passaged and replated multiple times, maintaining both GFP and RFP expression (not shown). Microscopic analysis revealed that many of the double-positive, fused cells exhibited two distinct nuclei (Supporting Information Fig. S1L–1S).
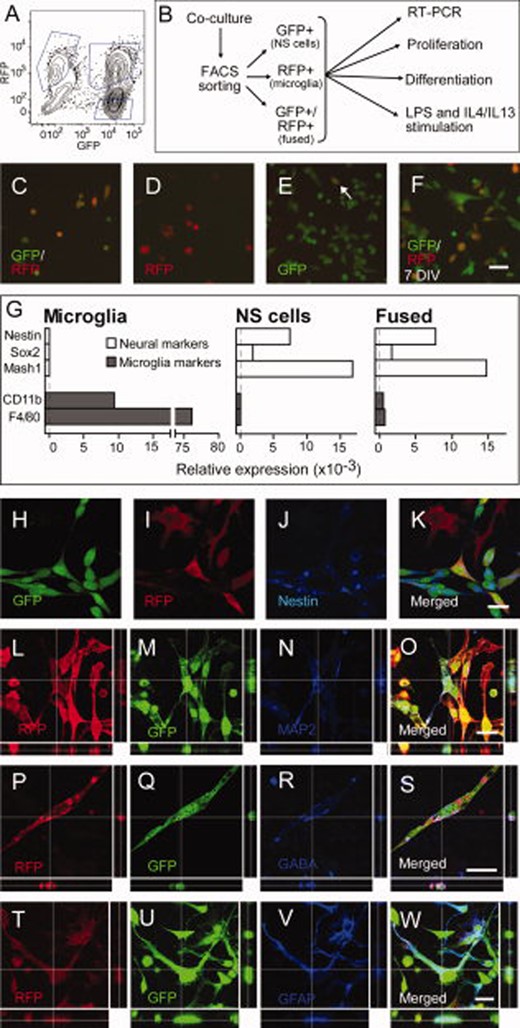
Sorted fused NS/microglia cells exhibit properties of both NS cells and microglia. (A): Example of FACS plot of a coculture; the gates represent the three populations (only GFP+, only RFP+, and double (GFP/RFP)-positive cells) that were sorted. (B): Schematic representation of the experimental design in these experiments. (C–E): Living cell images of the three fractions 6 hours after sorting; arrow in (E) depicts a GFP+/RFP+ cell. (F): GFP and RFP fluorescence in living cells sorted as double-positive, 7 days after sorting. (G): Expression of neuronal (nestin, Sox2 and Mash1) and microglial (CD11b and F4/80) genes in sorted microglia, NS cells, and fused cells. (H–K): Expression of nestin in cocultured NS cells and microglia. Note that microglia cells (I, only RFP+) are negative for nestin (J) while NS cells (H, only GFP+) and fused cells (K, GFP+/RFP+) are all immunoreactive for nestin (J). (L–O): Confocal images of fused GFP+/RFP+ cells expressing MAP2 after neuronal differentiation. Expression of GABA after neuronal differentiation (P–S) and GFAP after glial differentiation (T–W) of fused cells. Scale bars = 50 μm (F); 20 μm (K, O, S, W). Abbreviations: FACS, fluorescent-activated cell sorting; GFP, green fluorescent protein; GFAP, glial fibrillary acidic protein; NS, neural stem cell; RFP, red fluorescent protein; RT-PCR, reverse trandcriptase polymerase chain reaction; LPS, lipopolysaccharide; IL, interleukin.
Quantitative polymerase chain reaction (PCR) analysis of sorted GFP+/RFP+ cells showed that they expressed the neural markers, nestin, Sox2, and Mash1, similarly to NS cells (Fig. 3G). In addition, they expressed microglial markers CD11b and F4/80 (Fig. 3G), although at much lower levels as compared to nonfused microglia. Microglia markers were not expressed by NS cells and neural markers not by microglia (Fig. 3G).
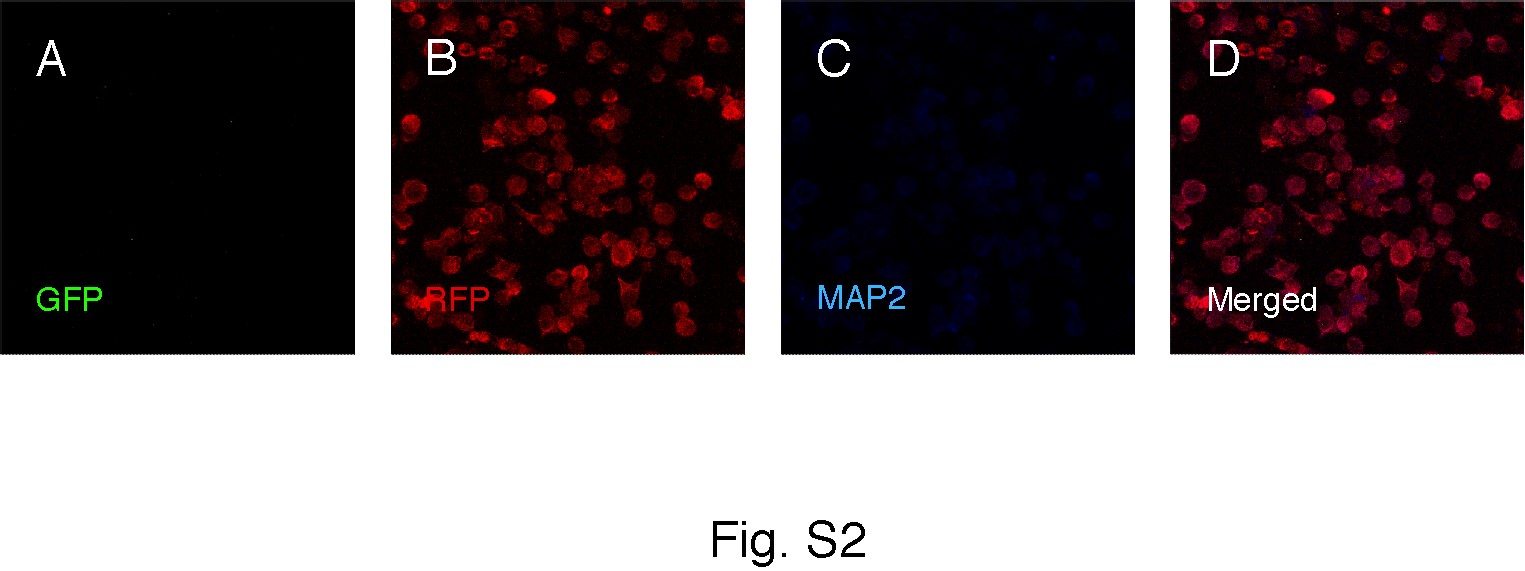
Under proliferative conditions, virtually all GFP+/RFP+ cells were also immunopositive for the NSC marker nestin (Fig. 3H–3K). We explored whether the fused cells retained the differentiation potential of NS cells. When applying a neuronal differentiation protocol to all three fractions of sorted cells, the GFP+/RFP+ fused cells and the GFP+ NS cells were efficiently differentiated to neurons, expressing MAP2 (Fig. 3L–3O), GABA (Fig. 3P–3S), βIII-tubulin, or doublecortin (DCX) (not shown). In contrast, the RFP+ microglia fraction did not give rise to any cells positive for neuronal markers (Supporting Information Fig. S2). Similarly, after 1 week of culturing using a glial differentiation protocol, the GFP+/RFP+ fused cells and the GFP+ NS cells but not the RFP+ microglia fraction gave rise to cells immunopositive for the astrocytic marker GFAP (Fig. 3T–3W).
Fused NS/Microglia Cells Display Functional Properties of NS Cells
Whole-cell patch-clamp recordings conducted on days 21, 24, and 27 of culturing NS cells demonstrated that they exhibit membrane properties similar to those previously described [3]. Depolarizing current injection frequently induced so-called “overshoot” action potentials (Supporting Information Fig. S3A), and in voltage-clamp mode, a large, depolarizing voltage-step induced a small inward current similar in shape and amplitude to those reported by Spiliotopoulos et al. [3]. The sorted fused cells recorded on days 21, 24, and 27 exhibited similar characteristics (“overshoot” action potentials and inward currents, Supporting Information Fig. S3B). However, the inward currents induced by a depolarizing voltage-step tended to be a magnitude larger than currents observed in NS cells, both here and by Spiliotopoulos et al. [3]. These electrophysiological characteristics are not typical of microglia [25, 26]. However, more detailed analysis of passive membrane properties showed that the fused cells tended to have lower input resistance than NS cells at all time points (Supplementary Table 1). Furthermore, at 24 and 27 days in vitro, the resting membrane potential of fused cells was significantly more hyperpolarized than that of NS cells. This pattern of alterations suggests that microglia may influence the membrane properties of the NS cells after fusion. Taken together, these results indicate that fused cells retain some membrane characteristics of NS cells but also exhibit alterations in passive membrane properties which may be derived from microglia.
Fused NS/Microglia Cells Also Display Functional Properties of Microglia
Microglia are activated when exposed to lipopolysaccharide (LPS) and increase their production of proinflammatory molecules such as inducible nitric oxide synthase (iNOS) and tumor necrosis factor (TNF)α [27]. These cells can also be alternatively activated with interleukin (IL)-4 and IL-13 and then express the anti-inflammatory markers arginase and CD206 [28]. We exposed the sorted double-positive cells to either LPS or IL-4/IL-13 for 24 hours. Quantitative PCR showed almost twofold increased expression of iNOS and TNF in response to LPS, and 8- and 53-fold increase of CD206 and arginase, respectively, following exposure to IL-4/IL-13 (Supporting Information Fig. S4A). These findings provided further evidence that fused NS/microglia cells retain some functional properties of microglia. However, the levels of expression of proinflammatory and anti-inflammatory molecules were much lower in fused cells as compared to nonfused microglia (Supporting Information Fig. S4B). NS cells did not express detectable levels of any of the examined proinflammatory or anti-inflammatory factors, either under normal culture conditions or following stimulation with LPS or IL-4/IL-13 (data not shown).
NS Cell and Microglia Fusion Involves a Phosphatidylserine-Dependent Mechanism
During myoblast [29] and macrophage [30] fusion as well as trophoblast formation [31], externalization and transient exposure of phosphatidylserine (PS) on the surface is an important event for the fusion of cells. We hypothesized that PS could also be involved in the fusion of NS cells and microglia. AnnexinV binds to exposed PS [32] and expression of the scavenger receptor CD36 is required on at least one of the fusing cells [33]. Staining with AnnexinV or CD36 antibody showed that NS cells constitutively expose PS and microglia express CD36 in our coculture system (Supporting Information Fig. S5).
In order to determine the functional significance of PS exposure on the membrane of NS cells for fusion with microglia, we masked the exposed PS with AnnexinV. In NS cultures treated with AnnexinV, the quantification of GFP+/RFP+ cells revealed a 50% decrease in the occurrence of fused cells (to 8.1% ± 1.1% from 15.4% ± 2.4% in nontreated controls), which supported an important role of PS in the fusion process.
Fused NS/Microglia Cells Fuse with Mouse Primary Cortical Cells In Vitro
Sorted fused NS/microglia cells were expanded in vitro and cocultured with CellVue-labeled mouse primary cortical cells. After 5 days of coculturing, cells were analyzed and sorted by flow cytometer to isolate GFP+/RFP+/CellVue+ triple-positive cells from the GFP+/RFP+ and the CellVue+ cell populations. The triple-positive population represented 2.7% of the total number of fused cells (Supporting Information Fig. S6A–S6B). Reanalysis of the sorted fractions confirmed efficiency of sorting to be 91.1% of double-positive cells out of which 97.1% were triple-positive (Supporting Information Fig. S6C–S6D).
For one set of experiments, the GFP+/RFP+/CellVue+ sorted cells were cultured in cortical differentiation medium for 1 week and subsequently processed for immunocytochemistry. The cells had developed elaborated long processes resembling mature neurons, and some of them were MAP2+ (Supporting Information Fig. S6E–S6H).
Microglia Fuse with Mature Neurons In Vitro and Their Activation Promotes This Process
To provide further evidence for a role of microglia in the fusion between NS cells and mature neurons, we cocultured rat fetal cortical cultures with GFP+ mouse microglia for 1–3 days. Already at 1 day after plating, we detected cells expressing both GFP and various neuronal markers such as MAP2, NeuN, and v-Glut1 (Fig. 4A–4K). Some of these cells were Iba1−, suggesting that not all fused cells retained microglia characteristics. We then investigated if the activation of microglia could influence the fusion between microglia and neurons. We applied LPS to the cocultures, analyzed them 3 days thereafter and found that LPS exposure increased the percentage of GFP+/MAP2+ cells by 87% compared to nontreated controls (Fig. 4L). We also explored whether IL-4, which is a macrophage fusion factor [34], could influence the fusion between microglia and NSCs. Mouse fetal cortex-derived GFP+ neurospheres were cocultured with mouse RFP+ microglia in the presence of either IL-4 or vehicle under adherent proliferative conditions. Treatment with IL-4 increased the percentage of fused GFP+/RFP+ cells 4.4- and 3.7-fold at 3 and 6 days in vitro, respectively (Fig. 4M).
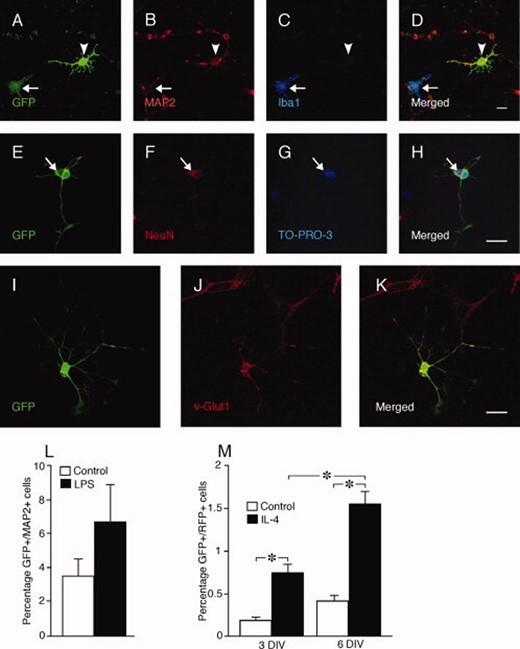
Microglia fuse with primary cortical neurons in vitro. (A–D): Coculturing rat fetal primary cortical cultures with GFP+ mouse microglia generates GFP+/MAP2+, Iba1− cells (arrowheads). Arrows indicate nonfused microglia. Fusion is confirmed by occurrence of binucleated neurons (NeuN+, arrow) also labeled with GFP (E–H), and of GFP+ cells expressing the neuronal marker v-Glut1 (I–K). (L): Number of fused cells differentiated to GFP+/MAP2+ neurons expressed as percentage of total number of Hoechst+, cells in cocultures of rat fetal primary cortical cultures and GFP+ mouse microglia, at 3 days treated with LPS or vehicle. (M): Number of fused GFP+/RFP+ cells expressed as percentage of total number of Hoechst+, cells in cocultures of mouse cortical GFP+ cells, expanded as neurospheres, and RFP+ microglia, at 3 and 6 days treated with IL-4 or vehicle. Means ± SEM. *, p < .05, unpaired t test (L) and one-way ANOVA and Dunnett's post hoc test (M). Scale bars = 20 μm. Abbreviations: GFP, green fluorescent protein; IL, interleukin; LPS, lipopolysaccharide.
Rodent Microglia and Fused NS/Microglia Cells Do Not Fuse with Human-Derived Cells In Vitro
We assessed if RFP+ microglia had the potential to fuse with human cortex-derived neurospheres and NS cells (both GFP+) as well as human iPS cell-derived lt-NES cells. However, after 3 and 5 days of coculturing, we did not detect any fused cells in any of the examined coculture conditions (Supporting Information Fig. S7A–S7H).
We also investigated the fusion capacity of the fused rodent NS/microglia cells with human-derived cells in vitro. Sorted fused GFP+/RFP+ cells were cocultured with human CellVue-labeled lt-NES cells and fetal cortex-derived primary cells which had been cultured for 20 days. After 3 and 5 days of coculture, no triple-labeled GFP+/RFP+/CellVue+ cells were detected. The vast majority of the cortical cells exhibited morphological characteristics of neurons and were immunoreactive to the cortical marker v-Glut1. After 4–5 days of coculturing, cells were stained for GFP, RFP, and v-Glut1. When only fused cells were cultured for 5 days in cortical differentiation medium, they were immune-negative for v-Glut1. We performed extensive confocal analysis of the cocultured cells, which were stained for GFP, RFP, v-Glut1, and the human cytoplasmic marker SC121. The fused cells were located in close contact with the cortical cells but we did not find any examples of GFP+/RFP+/v-Glut+, or SC121+ cells (Supporting Information Fig. S7I–S7L).
Transplanted NS Cells Fuse with Mature Neurons In Vivo
We implanted GFP+ mouse NS cells in the cerebral cortex of newborn rat pups. At 1 week after transplantation, all GFP+ cells had immature morphology (data not shown). Three weeks later two distinct populations of GFP+ cells were observed (Fig. 5A–5D). One group of cells with immature morphology was located in superficial cortical layers, radiating from the transplant core in a ribbon-like fashion. The other group with characteristics of mature cortical pyramidal neurons represented 55.0% ± 18.8% of all GFP+ cells, was located mostly in cortical layer V, had very long apical dendrites, and expressed the mature neuronal marker NeuN (Fig. 5E–5H). Similar results were observed when GFP+ or RFP+ NS cells were implanted into cerebral cortex of mouse pups. Mature GFP+ or RFP+ neurons were detected from 4 weeks after transplantation, but they were much fewer in mice compared to rats (2.0% ± 0.8% of total number of GFP+ or RFP+ cells). Also in mice, the GFP+ or RFP+ pyramidal-like cells were located in layer V and had mature dendritic arborizations with spines (Fig. 5I–5L). Few (about 1%) GFP+ cells were positive for the microglia marker Iba1 (Fig. 5M–5P).
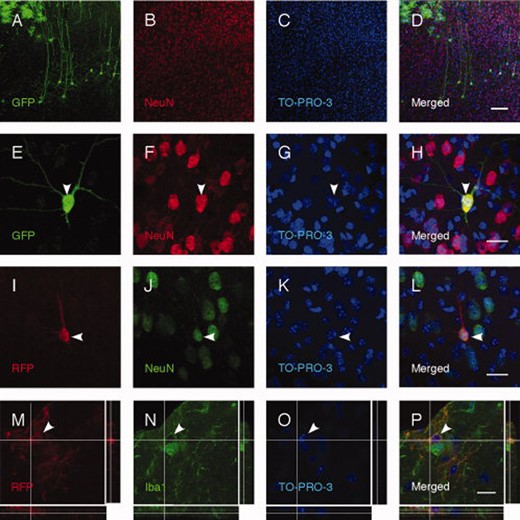
Transplanted NS cells remain undifferentiated or acquire properties of mature cortical neurons in vivo. (A–H): More than half of transplanted GFP+ NS cells in rat neonates display a cortical pyramidal neuron morphology and are NeuN positive. Similar results are obtained after transplantation of RFP+ NS cells in mouse neonates (I–L). (M–P): Some RFP+ NS cells grafted into mouse neonates express the microglia marker Iba1. Arrowheads indicate double-labeled cells. Scale bars = 100 μm (D), 20 μm (H, L, and P). Abbreviations: GFP, green fluorescent protein; RFP, RFP.
Based on our in vitro data, we hypothesized that the occurrence of mature, GFP+ or RFP+, fully differentiated neurons at 4 weeks was due to fusion between grafted NS cells and host mature neurons. To support this hypothesis, we used two species-specific antibodies, M2 and M6, which recognize membrane antigens on neurons and glia of mouse origin [35, 36]. Immunostaining with M2/M6 antibodies of brain sections from rats grafted with mouse ES cell-derived NS cells revealed that virtually all GFP+ cells with mature pyramidal neuron morphology were negative for the mouse-specific neuronal and glial markers. This finding indicated that these cells were of rat (host) rather than mouse (donor) origin (Fig. 6A–6D). Other cells in the graft with immature morphology were labeled with the M2/M6 markers, confirming the efficacy and specificity of the staining. Similar to what was observed in vitro, some of the M2/M6 immunoreactive cells were also positive for the microglia marker Iba1 (Fig. 6E–6H), although M2/M6 antibodies do not stain mouse microglia (data not shown).
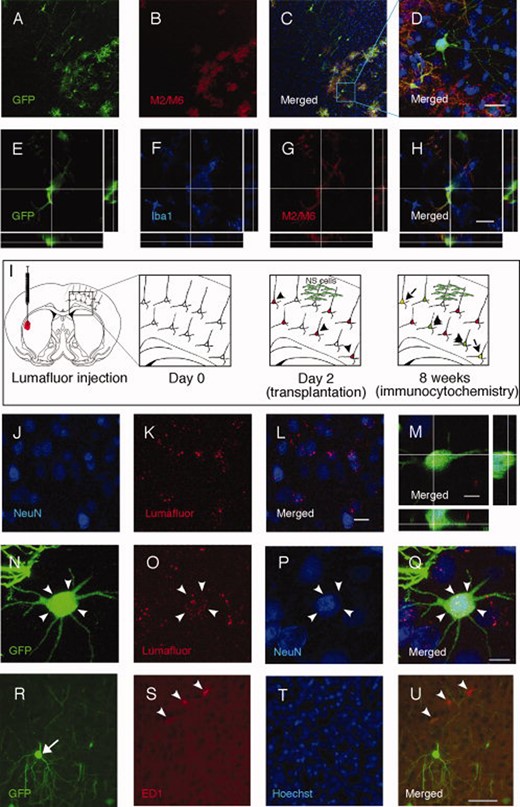
Transplanted neural stem (NS) cells fuse with mature cortical neurons in vivo. (A–D): Mouse embryonic stem (ES) cell-derived GFP+ NS cells displaying morphology of mature cortical neurons after intracortical transplantation into neonatal rat brain do not stain with mouse-specific M2/M6 antibodies in contrast to undifferentiated cells. (E–H): Some M2/M6-positive cells express the microglial marker Iba1. (I–L): Cortical neurons labeled with fluorescent retrograde tracer Lumafluor injected in contralateral striatum. (M): Confocal image with orthogonal view of the grafted GFP+ NS cell expressing Lumafluor and stained with NeuN antibody. (N–Q): Confocal image of the cell shown in M and depicted with arrow heads. (R–U): Grafted GFP+NS cells (arrow) located in close proximity to activated ED1+ microglia (arrow heads). Scale bars = 20 μm (D, H, and L), 10 μm (M), 5 μm (Q), 50 μm (U). Abbreviation: GFP, green fluorescent protein.
To provide further evidence for host origin of the GFP+ or RFP+ cells with mature neuronal morphology, we prelabeled cortical pyramidal neurons in the host brain by injecting a retrograde tracer (Lumafluor RedBeads) in the dorsolateral striatum in 2 days old mouse pups (Fig. 6I). This treatment resulted in fluorescent labeling of several brain areas including pyramidal neurons in the contralateral cortex. After 2 days, when the retrograde transport of the label was completed, GFP+ NS cells were implanted into the prelabeled cortex (Fig. 6I). Animals were allowed to survive for 8 weeks. We detected both Lumafluor and GFP in some pyramidal neurons, which indicated that they were of host origin and had not differentiated from grafted NS cells (Fig. 6J–6Q). Grafted GFP+ NS cells were often located in close proximity to activated ED1+ microglia (Fig. 6R–6U).
When cells have fused, such as BMDCs and cerebellar Purkinje neurons, two nuclei are occasionally detected [37]. We carefully analyzed confocal images of GFP+ pyramidal neurons in brain sections from rats which had been grafted with mouse NS cells. We found that 17.2% of the analyzed cells (10 out of 58 in three different animals) had two distinct nuclei (Fig. 7A; Supporting Information Fig. S8). In some cases, the supernumerary nucleus was located on the apical dendrite (Fig. 7B–7D). Interestingly, not only the pyramidal neurons but also, more rarely, some of the other cells in the graft, notably M2/M6 positive, had two nuclei (Fig. 7E–7I).
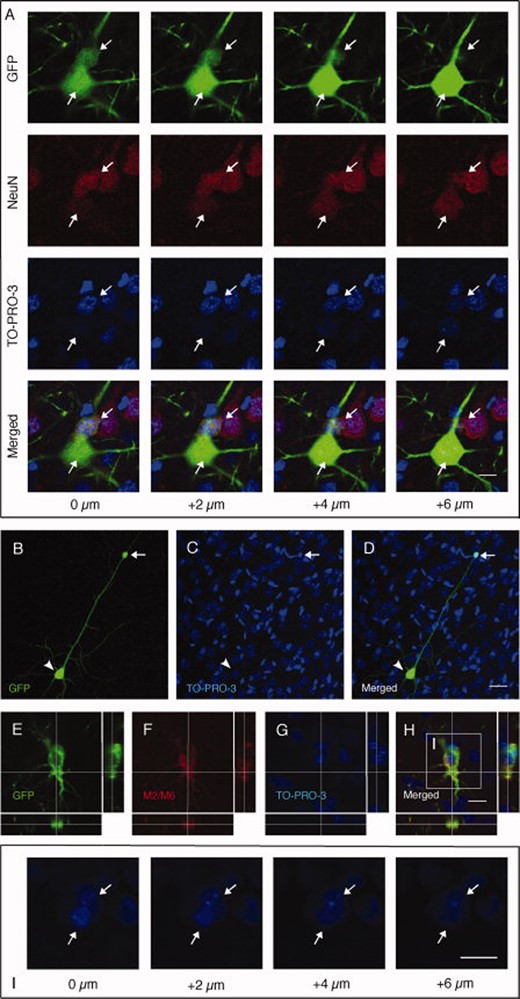
Grafted neural stem (NS) cells form heterokaryons. (A): Confocal reconstruction of GFP+ pyramidal neuron stained with NeuN and TO-PRO-3 and containing two distinct nuclei (arrows) from the cortex of a rat grafted with GFP+ NS cells. Every column represents one focal plane 2 μm apart from the previous one. (B–D): GFP+ pyramidal neuron (arrowhead) carrying one supernumerary nucleus (arrow) on the apical dendrite. (E-H): GFP and M2/M6 double-positive cell which contains two nuclei as visualized by TO-PRO-3 staining and shown in different confocal planes (I). Scale bars = 10 μm (A, H, and I), 50 μm (D). Abbreviation: GFP, green fluorescent protein.
Discussion
Here we describe, for the first time, fusion of ES cell-derived NS cells with mature neurons. We present in vitro evidence that the NS cells fuse with microglia, at least partly, through interaction between PS exposed on the surface of NS cells and CD36 receptor on microglia. The fused NS/microglia cells are able to fuse with primary neurons. Following intracerebral transplantation, the NS cells fuse with rat cortical neurons. Whether the NS cells first fuse with microglia and these chimeras then fuse with mature neurons in vivo similar to what was observed in vitro remains to be determined. It is possible that the activation of microglia/macrophages caused by the transplantation procedure is involved in the fusion process.
The occurrence of fusion between NS cells and microglia was inferred from several observations. First, coculturing of mouse or rat microglia with mouse NS cells, labeled with different fluorescent markers, lead to the appearance of many cells bearing two markers, indicating the formation of a hybrid cell. Second, to exclude transfer of fluorescent proteins between cells, we prelabeled GFP+ microglia with BrdU before coculturing them with RFP+ NS cells. The occurrence of GFP+/RFP+/BrdU+ triple-positive cells provides further evidence that microglia and NS cells had fused. Third, to exclude that dead NS cells had been engulfed by microglia, thereby causing double-labeling, we performed a set of cocultures of microglia with NS cells killed by freezing–thawing. The absence of any fused cells in these cultures indicated that fusion required living NS cells and further excluded the possibility of nonspecific label transfer. Fourth, also in living cocultures it was possible to observe healthy-looking cells exhibiting double-fluorescence. These findings added to the body of evidence that fusion could be observed regardless of the detection method used. Fifth, the presumed fusion in vitro was a spontaneous and continuous process, and the number of double-positive cells increased over time. Finally, fusion was not only confined to ES cell-derived NS cells but also observed in cocultures of microglia with mouse fetal cortex-derived NS cells (Cor1). Taken together, our data strongly indicate that NS cell–microglia fusion, as we have observed it, is not an artifact but due to specific interaction between these cell types. Interestingly, we observed no fusion after coculturing rodent microglia with cells of human origin, that is, fetal cortex derived-NS cells or neurospheres, or iPS cell-derived lt-NES cells. This finding suggests that microglia fusion is a species-sensitive phenomenon.
The fused NS/microglia cells maintained the expression of the neural markers nestin, Sox2, and Mash1 and, in addition, expressed low levels of the microglia markers CD11b and F4/80. Microglia markers are not expressed by NS cells and neural markers not by microglia. The fused cells were able to differentiate into both neurons and astrocytes. Importantly, we found that the fused cells exhibited functional properties of both NS cells and to some extent also of microglia. First, the electrophysiological characteristics of the fused cells resembled those of NS cells although they were not identical. The observed changes in the amplitude of inward currents, lower input resistances, and more hyperpolarized resting membrane potential suggest that the fusion with microglia influences the membrane properties of the NS cells. Second, the fused cells increased their expression of proinflammatory molecules when activated with LPS and of anti-inflammatory molecules when stimulated with IL-4/IL-13. No such change in expression of pro inflammatory or anti-inflammatory molecules was detected in NS cells following LPS or IL-4/IL-13 exposure, respectively. Our findings suggest that the fused cells can partially exhibit the phenotype of both classically activated M1 macrophages, which are involved in neurotoxicity, and alternatively activated M2 macrophages, which are neuroprotective.
It is well-established that macrophages can fuse to each other, and that multinucleated giant cells originating from fusion of macrophages are recruited to the granulomatous site [38]. Interestingly, fusion of macrophages is induced by IL-4 and granulocyte macrophage colony-stimulating factor. Exposure of PS on the cell surface and lipid recognition by scavenger receptor CD36 are required for this event [33]. In analogy, we found here that PS is exposed on NS cells and that microglia express CD36, which could be involved in the fusion process. Normally, PS exposure on apoptotic cells is considered as a signal to macrophages for their clearance [39]. Our data indicate that NS cells expose PS without undergoing apoptosis and that the fusion of microglia with NS cells does not lead to removal of NS cells but to formation of a new chimeric cell, bearing the properties of both parental cells. These fused cells can be passaged multiple times without loosing their dual, neural and microglial properties.
Spontaneous fusion of NSCs, derived from mouse fetal forebrain or adult SVZ, with ES cells has been demonstrated in vitro [13], the hybrid cells maintaining properties of ES cells including self-renewal capacity, and pluripotency, and ability to efficiently differentiate to cells of different lineages. Also BMDCs fuse spontaneously with ES cells [40] and NSCs [41] in vitro. In BMDCs fused with ES cells, the properties of ES cells are dominant: the undifferentiated fused cells express Oct3/4 and UTF1 and when differentiated, express markers of mesoderm, endoderm, and ectoderm. If the fused cells are cultured on PA6 cells, they predominantly differentiate to morphologically neuron-like cells, expressing tyrosine hydroxylase, and when injected into immunocompromised mice, form teratomas. In contrast to the hybrid cells between ES cells and either NSCs or BMDCs, our data show that fused NS and microglia cells are chimeras exhibiting the properties of both parental cells.
Under proliferating conditions, approximately 0.2% of NSCs isolated from adult rodent brain could fuse to each other [15]. Fused cells did not proliferate and could not be propagated, suggesting that they were not viable. In contrast, the chimeric cells derived from NS cells and microglia in our study proliferated, could be passaged multiple times (they could also be frozen/thawed and recultured), and efficiently differentiated to neurons and astrocytes.
About 15% of NSCs isolated from cerebellum and SVZ of early postnatal mice also fuse spontaneously [14]. By using PU.1 knockout mice which lack myeloid progenitors and therefore microglia and macrophages, the authors concluded that microglia are not important for fusion of NSCs to occur. In contrast, our in vitro data indicated that the fusion capacity of ES cell-derived NS cells is dependent on the presence of microglia in the culture. It must be noted, although, that Chen et al. [14] used neurosphere cultures and not pure microglia cultures. The discrepancies compared to our results are probably due to the fact that microglia were rare in their cultures, and the phenomenon these authors observed was different from that described here.
Several lines of evidence indicate that ES cell-derived NS cells fuse with mature host neurons in vivo after transplantation. First, virtually all GFP+ and RFP+ cells with mature pyramidal neuron morphology were negative for the mouse-specific markers (M2/M6) in brain sections from rats grafted with mouse ES cell-derived GFP+ NS cells. Second, after transplantation of GFP+ NS cells into cortex where pyramidal neurons had been prelabeled retrogradely with a fluorescent tracer, we detected colabeling of tracer and GFP in some pyramidal neurons, indicating that they were of host origin. Third, confocal analysis of GFP+ pyramidal neurons in brain sections from rats grafted with NS cells revealed that 17.2% of the cells had two distinct nuclei in their soma.
BMDCs from genetically modified mice which were implanted in lethally irradiated mice fused with cardiomyocytes, hepatocytes, and Purkinje neurons [11, 12, 41]. Interestingly, Nygren et al. [12] demonstrated that blood cells not only of myeloid but also of lymphoid lineage can form hybrids but almost exclusively in response to injury or inflammation. Thus, activation of microglia/macrophages seems to be important for the fusion process. We observed that grafted cells that have fused are often in the vicinity of activated microglia. Although we do not have any direct in vivo evidence that this activation of microglia is involved in the fusion of grafted NS cells with resident cortical neurons, our in vitro data provide supportive evidence that this could indeed be the case. Removal of microglia from cultures of NS cells and primary cortical neurons substantially decreased the number of fused cells. This was reversed by the reintroduction of microglia after the Mac1-saporin treatment, strongly indicating that microglia were necessary for the fusion to occur. Fused NS/microglia cells were able to fuse with primary cortical cells. Fusion between microglia and fetal cortical neurons or NSCs in vitro was enhanced by LPS-induced activation of microglia or treatment with IL-4, respectively. In line with these in vitro data, we observed in vivo that some grafted RFP+ NS or M2/M6+ (markers which under normal conditions do not stain microglia) cells were also positive for the microglia marker Iba1.
The main observation here that transplanted NS cells fuse with a substantial number of mature neurons in the intact brain raises important issues. First, if microglia are involved in mediating the fusion process in vivo, it adds an additional fusogenic role of microglia to their other modes of action after NSC transplantation. It is plausible that this role is even more important after transplantation into brains affected by acute or chronic neurodenerative disorders associated with microglia activation. Our findings also suggest that the characteristics of the pathological environment, whether it is pro inflammatory or anti-inflammatory, will determine which microglia properties will dominate in the hybrids. Second, although the functional role of fusion remains unknown, the formation of hybrids could hypothetically be beneficial and contribute to the improved functional recovery after NSC transplantation in brain disease. If this is the case, the fusion process may become a novel therapeutic target and provide a method for generating chimeric cells with defined properties such as releasing factors supporting the functional integrity of the injured and diseased brain.
Conclusions
In the present work, we show that ES-derived neural stem cells fuse with microglia and neurons both in vitro and in vivo. We also demonstrate that microglia are responsible for mediating the fusion between NS cells and neurons, in a process that is facilitated by activation. This phenomenon is of potential relevance to interpret xenograft studies and understand the true action of NSCs once implanted. Moreover, it highlights the multifaceted role of microglia and their activation in the both healthy and diseased central nervous system.
Acknowledgements
We thank Elena Cattaneo and Luciano Conti for sharing NS cells and for useful discussions in the early stages of the work, and Teona Roschupkina for help with cell sorting. This work was supported by grants from Swedish Research Council, European Union projects StemStroke (037526) and TargetBraIn (279017), AFA Foundation, and Swedish Government Initiative for Strategic Research Areas (StemTherapy). C.C. was partially supported by a grant from Giuseppina Mai Foundation.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
Author notes
Author contributions: C.C. and E.M.: conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing, H.A., J.W., and J.C.B.: collection and assembly of data and data analysis and interpretation; O.L. and Z.K.: conception and design, financial support, data analysis and interpretation, and manuscript writing. C.C. and E.M. contributed equally to this article.
Disclosure of potential conflicts of interest is found at the end of this article.
First published online in STEM CELLSEXPRESS November 7, 2012.
Telephone: +46 46 222 0276; Fax: +46 46 222 0560
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}