

Abstract
Healthy synovium is critical for joint homeostasis. Synovial inflammation (synovitis) is implicated in the onset, progression and symptomatic presentation of arthritic joint diseases such as rheumatoid arthritis and osteoarthritis. Thus, the synovium is a promising target for the development of novel, disease-modifying therapeutics. However, target exploration is hampered by a lack of good pre-clinical models that accurately replicate human physiology and that are developed in a way that allows for widespread uptake. The current study presents a multi-channel, microfluidic, organ-on-a-chip (OOAC) model, comprising a 3D configuration of the human synovium and its associated vasculature, with biomechanical and inflammatory stimulation, built upon a commercially available OOAC platform. Healthy human fibroblast-like synoviocytes (hFLS) were co-cultured with human umbilical vein endothelial cells (HUVECs) with appropriate matrix proteins, separated by a flexible, porous membrane. The model was developed within the Emulate organ-chip platform enabling the application of physiological biomechanical stimulation in the form of fluid shear and cyclic tensile strain. The hFLS exhibited characteristic morphology, cytoskeletal architecture and matrix protein deposition. Synovial inflammation was initiated through the addition of interleukin−1β (IL−1β) into the synovium channel resulting in the increased secretion of inflammatory and catabolic mediators, interleukin-6 (IL−6), prostaglandin E2 (PGE2), matrix metalloproteinase 1 (MMP−1), as well as the synovial fluid constituent protein, hyaluronan. Enhanced expression of the inflammatory marker, intercellular adhesion molecule-1 (ICAM-1), was observed in HUVECs in the vascular channel, accompanied by increased attachment of circulating monocytes. This vascularised human synovium-on-a-chip model recapitulates a number of the functional characteristics of both healthy and inflamed human synovium. Thus, this model offers the first human synovium organ-chip suitable for widespread adoption to understand synovial joint disease mechanisms, permit the identification of novel therapeutic targets and support pre-clinical testing of therapies.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The human synovium is a membrane-like structure that lines the inner surfaces of diarthrodial joints and, under normal conditions, plays several important roles in maintaining joint homeostasis [1]. Dysregulation of synovial function is implicated in the onset and progression of arthritic joint diseases, in which synovial inflammation (synovitis) is common. The synovium is the primary site of pathology in inflammatory arthridites, such as rheumatoid arthritis (RA), with synovitis a ubiquitous feature that is widely appreciated to be the driving factor of secondary damage to the adjacent cartilage and bone [2, 3]. There is also increasing interest in the contribution of synovitis to the pathogenesis of osteoarthritis (OA), traditionally considered a non-inflammatory, degenerative disease. It has been reported that chronic, low-level synovial inflammation plays a role in OA pathogenesis in at least a subset of patients and is associated with clinical disease features such as cartilage loss, joint space narrowing and pain [4–7].
Synovitis is characterised by tissue thickening, cellular hyperplasia, and immune cell infiltration, along with the increased release of proinflammatory mediators such as cytokines, nitric oxide (NO) and prostaglandin E2 (PGE2), by the resident synovial cells [8]. These factors attract circulating immune cells and promote angiogenesis (for review see Sellam and Berenbaum, 2010 [9]; figure 1(A)). Additionally, there is an increase in the secretion of proteolytic enzymes, such as matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), which disrupts the delicate balance between catabolism and repair in the joint and contributes to cartilage degeneration and bone remodelling. In turn, the products of cartilage degradation further amplify synovial inflammation, creating a vicious circle that, if left untreated, eventually leads to joint destruction [10]. Thus, synovium-targeted therapies hold great potential for the treatment of joint diseases. Pre-clinical models of the human synovium are required to understand disease processes and enable target identification for the development of novel, disease-modifying therapeutics.
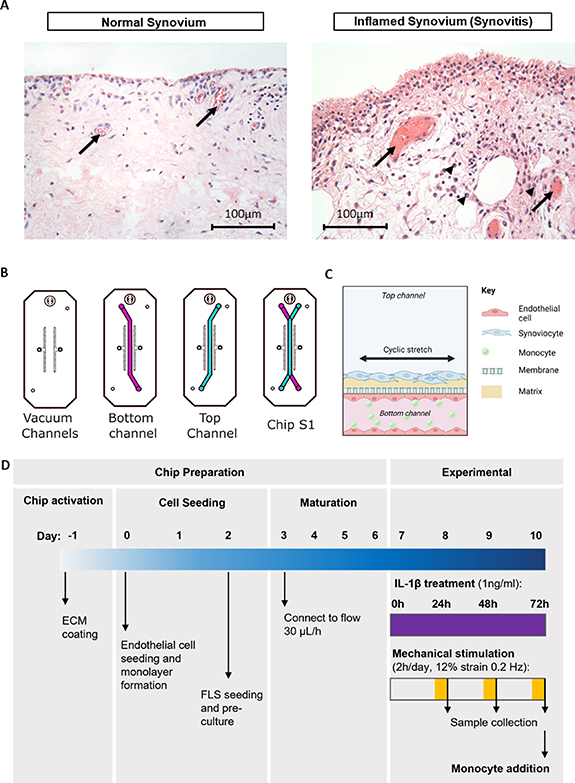
Figure 1. Developing an organ-on-a-chip model of vascularised synovium. (A) Representative histological images of normal synovium (left) and inflamed (high-grade synovitis) synovium (right) as determined by the Krenn synovitis score [20], with associated blood vessels (arrows). The inflamed synovium demonstrates intimal layer thickening, increased stromal cellularity and the presence of macrophages (arrow heads) in proximity to blood vessels, suggesting increased monocyte extravasation. (B) Schematic of the Chip S1® from Emulate Inc., comprising two overlapping channels separated by a permeable membrane. Top channel (blue) is 1 mm in height, bottom channel (pink) is 0.2 mm in height. The channels are flanked by two vacuum channels which are used to apply cyclic tensile strain to the co-culture region. (C) Schematic (created using Biorender.com) illustrating the final synovium-on-a-chip model with human fibroblast-like synoviocytes (hFLSs) cultured in the top channel, human umbilical vein endothelial cells (HUVECs) cultured in the bottom channel in co-culture with THP-1 monocytes added under flow. (D) Schematic highlighting the key steps for chip preparation and subsequent experimentation whereby chips are subjected to interleukin−1β (IL−1β) stimulation in the presence of mechanical stimulation (2 h, 12% at 0.2 Hz).
Download figure:
Standard image High-resolution imageTraditional systems used for pre-clinical drug development rely heavily on simple in vitro models that lack complexity, and animal models that are limited in terms of reproducibility and translatability [11]. These systems are inefficient due to their common lack of relevance to human physiology and, consequently, the current rate of attrition for new drugs at the pre-clinical stage is suggested to be in the region of 90% [12]. Organ-on-a-chip (OOAC) systems aim to overcome these shortfalls by creating improved predictive in vitro models of tissue function that more accurately replicate in vivo physiological responses of the organ and its interactions with surrounding tissues. To achieve this, OOACs incorporate key components of the extracellular microenvironment that regulate cellular phenotype and function, such as extracellular matrix (ECM), tissue organisation, tissue stiffness, mechanical stimulation and adjacent tissue interactions such as vasculature to facilitate nutrient delivery, drug delivery or examine disease processes such as immune cell recruitment.
Several groups have utilised OOAC technology to generate in vitro mimics of the synovium, utilising a range of cell sources and configurations for a number of different purposes [2, 13–15]. Of particular interest, Mondadori and colleagues recently reported on the development of a multi-channel OOAC system to study the process of monocyte extravasation into the synovium, using human cells. This elegant system comprised both a vascularised synovium compartment and a cartilage compartment, with the two separated by an additional channel designed for synovial fluid (SF) [14]. This model demonstrated the capability to mimic a number of important interactions in the human joint and was convincingly used by the group to demonstrate monocyte extravasation in response to both chemokines and OA SF. While each of the aforementioned models has been developed with a specific purpose and have been used successfully to address their respective research questions, they share common limitations that may restrict their widespread utility for the discovery of disease modifying therapeutics. Firstly, each of these existing models has been developed on in-house generated OOAC systems, which, while allowing for increased configurational freedom, limits their uptake by other academic and industrial users. Secondly, these models commonly lack physiologically relevant biomechanical stimulation. In vivo, the synovium is a deformable surface between the other joint tissues which is able to fold, roll and stretch during locomotion, allowing for the movement of less deformable tissues [1]. Thus, the synovium is subjected to biomechanical stimulation not only in the form of shear, due to the movement of SF, but also strain. Therefore, it is important that biomimetic models of the human synovium incorporate such mechanical loading capabilities. In previous in vitro work, cyclic tensile strain and fluid shear have been shown to independently modulate metabolic and inflammatory responses in synovial explants [16] and isolated FLS [17–19], but have not previously been applied simultaneously, as in our model.
In the present study, we report the first vascularised, synovium-on-a-chip model with mechanical stimulation, which has been developed to study the healthy synovium, as well as to investigate synovial inflammation and its effect on circulating monocyte behaviour. The model was developed upon a commercially available platform that allows for widespread adoption without the need for specialist knowledge of device design and fabrication. A number of 2D cell culture experiments were initially carried out to optimise culture and experimental conditions, which were subsequently applied to the OOAC model. The model comprised primary human fibroblast-like synoviocytes (hFLS) that replicate the synovial lining and its close association with the vasculature, which itself was recreated using human umbilical vein endothelial cells (HUVECs). Physiological mechanical stimulation was applied in the form of both fluid shear and periodic cyclic tensile strain. We demonstrated that this optimised organ-chip model can be used to examine inflammation and the first stage of monocyte extravasation; attachment to the endothelium.
2. Materials and methods
2.1. Cell culture
Healthy human fibroblast like synoviocytes (hFLS) from 3 male donors aged 32, 39 and 75 were purchased from a commercial source (Articular Engineering, Illinois, USA) and used from passage 1-6 for experiments. Cells were cultured in monolayer in Dulbecco's modified eagle medium: nutrient mixture F-12 (DMEM/F12; 1:1) supplemented with 2 mM penicillin/streptomycin and 10% (v/v) foetal bovine serum (FBS; all Gibco, Loughborough, UK). hFLS were cultured to 80% confluence prior to passaging and seeding for experiments. HUVECs (Lonza, Cambridge, UK) were used from passage 3–7 and cultured in endothelial growth medium-2 (EGM-2; Lonza). THP-1 monocytes (Invivogen, Toulouse, France) were cultured in Roswell Park Memorial Institute (RPMI) medium (Gibco) supplemented with 2 mM penicillin/streptomycin and 10% (v/v) FBS. All cells were cultured in a humidified incubator at 37 °C with 5% CO2.
2.2. Preparation and establishment of the vascularised synovium-on-a-chip
The Chip-S1® (Emulate Inc., Boston, USA) was used (figure 1(B)) to generate the vascularised synovium-on-a-chip model. This PDMS based chip comprises two overlapping channels separated by a thin (50 µm), porous (7 µm pore diameter) membrane. The channels are 1 mm in width and the channel height is 0.2 mm and 1 mm for the bottom and top channels respectively. Each channel is connected to its own medium reservoir using the Pod™ system (Emulate) such that each cell population can be supplied individually with required media formulations, and fluid shear can be applied to each channel independently. Two vacuum channels either side of the culture channels are used to apply uniaxial cyclic tensile strain across the membrane (figure 1(B)). A commercially available organ-chip system was selected for use in the present study to allow for the wider research community to adopt the optimised model without the need for specialist knowledge of chip fabrication techniques.
Prior to cell seeding, the inner surfaces of the channel were activated according to standard protocols (emulatebio.com; 'Basic Research Kit Protocol: EP223). In brief, 0.5 mg ml−1 ER-1 solution (Emulate) was added to each channel and the chips were then treated with UV light for 10 min. The channels were aspirated, and the process repeated with additional ER-1 solution. Channels were then washed with ER-2 solution (Emulate) followed by phosphate buffered saline (PBS; Gibco). Extracellular matrix, in the form of 500 µg ml−1 collagen Type I (Corning, Arizona, USA), was added to the top channel, and a mix of 250 µg ml−1 collagen type 1 and 25 µg ml−1 fibronectin (Thermo Fisher Scientific, Massachusetts, USA) added to the bottom channel. Chips were incubated overnight at 4 °C, followed by 1 h at 37 °C.
In the final chip configuration, hFLS were cultured in the top channel and HUVECs in the bottom channel (see schematic representation in figure 1(C)). Following the matrix coating of the two channels, chips were washed with pre-warmed culture medium and HUVECs seeded to the bottom channel at a concentration of 6 × 106 cells ml−1. Chips were inverted for 2 h to encourage attachment to the membrane, then righted and cultured for a further 24–48 h to encourage formation of a robust monolayer with associated tight junctions. hFLS were then seeded to the top channel at a concentration of 1 × 106cells ml−1 and allowed to attach for 4 h prior to gentle washing. After a further 24 h, chips were connected to the automated culture module (ZÖE; Emulate) and cultured with their respective media flowing at a rate of 30 µl h−1 for 7 d to encourage deposition of hFLS-derived matrix before the experimental parameters, optimised as described subsequently, were applied.
2.3. 2D optimisation of synovium-on-a-chip experimental parameters
Several experiments were initially carried out on 2D monocultures of hFLS to optimise experimental parameters, which were subsequently applied to the organ-chip system. Firstly, an interleukin−1β (IL−1β) dose response experiment was performed. hFLS were subjected to an IL−1β (Peprotech, London, UK) dose of 0, 0.1, 1 or 10 ng ml−1 for 24 h to identify an optimal dose for use in subsequent studies, with culture supernatant examined for the presence of inflammatory signalling molecules (NO, PGE2, interleukin-6 (IL−6) and matrix metalloproteinase 1 (MMP-1)). Based on results from these experiments (detailed in the results section) a dose of 1 ng ml−1 of IL−1β was used for subsequent experiments.
Mechanical strain regimes were also initially optimised off-chip. Cyclic tensile strain (CTS) was applied using a commercially available stretch system (Flexcell; Flexcell® International Corporation, North Carolina, USA). hFLS were seeded on Collagen Type I coated Uniflex plates (UF4001-C, Dunn Labortechnik, Thelenberg, Germany). Plates were additionally coated overnight with 50 µg ml−1 Collagen type 1 (354249; Corning, Arizona, USA) at 4 °C followed by incubation for 1 h at 37 °C and washing with culture medium. hFLS were seeded at a density of 5000 cells cm−2 and cultured until confluent (approx. 7 d). hFLS were subjected to a strain regime which comprised 22 h of unloading immediately followed by 2 h of loading, at 2% or 12% strain at a frequency of 0.2 Hz using a sinusoidal waveform. These experiments were performed in the presence or absence of 1 ng ml−1 IL−1β.
2.4. Synovium-on-a-chip experimental procedure
The optimised mechanical regime was applied to the chips from day 7, following the initial culture period, such that chips were unstretched for 22 h then subjected to cyclic stretch for 2 h at 12% strain, 0.2 Hz using a sinusoidal waveform. This was repeated for 3 d (figure 1(D)). At the same time, inflammation was induced in the synovium-on-a-chip through addition of the optimised dose of 1 ng ml−1 IL−1β in serum-free hFLS media to the top channel inlet media reservoir at day 7. IL−1β containing media was then flowed through the top channel at a rate of 60 µl h−1, for 3 d (figure 1(D)).
2.5. THP-1 monocyte recruitment assay
On day 10, THP-1 monocytes were labelled with 2 µM cell tracker dye (CMRA orange, Thermo Fisher Scientific) for 15 min in serum-free medium at 37 °C. Cells were then centrifuged at 200 ×g for 8 min to form a pellet, washed with serum-free medium then resuspended to a concentration of 4 × 106 cells ml−1. Cells were subsequently mixed 1:1 with an equal volume of buoyancy medium (1.6% v/v gelzan in Percoll; Sigma-Aldrich, Dorset, UK) and added to the inlet reservoir of the bottom channel of the Pod™. Fluorescently labelled monocytes were flushed through the bottom channel at 1000 µl h−1 for 10 min then incubated under static conditions for 3 h. The channel was then flushed with fresh EGM-2 for 10 min to removed unattached cells and chips were imaged to assess monocyte recruitment using confocal microscopy. Labelled monocytes were counted using Image J (Image J 1.53c, National Institutes of Health, USA) for 15 fields of view per chip.
2.6. Endpoint analyses
2.6.1. Sample collection
At the end of 2D cell culture experiments, cells were immediately fixed with 4% paraformaldehyde (PFA; Thermo Fisher Scientific) for 10 min for histological analysis, or lysed using buffer RLT (Qiagen, Manchester, UK) supplemented with 0.2% Dithiothreitol (Sigma-Aldrich). Cell lysates were stored at −80 °C for subsequent RNA extraction and gene expression analyses. Cell culture media was collected from 2D cultures as per standard protocols. For effluent collection from synovium-on-a-chip, the Pods™ containing the synovium chips were temporarily disconnected from the ZÖE and media samples collected from the outlet reservoir for each channel separately. For both 2D and chip experiments, conditioned media was centrifuged at 220 ×g for 10 min, and the supernatant aliquoted and stored at −80 °C for downstream analyses.
2.6.2. RNA isolation, cDNA synthesis and qRT-PCR
Total RNA was extracted from cell lysates using the RNeasy mini kit (Qiagen) according to the manufacturer's instructions. RNA was re-suspended in RNase-free water, and yields and indicative purity were confirmed using Nanodrop™ (Thermo Fisher Scientific). For Flexcell studies, 250 ng RNA was used to synthesise cDNA and reverse transcription was performed using the Quantitect cDNA synthesis kit (Qiagen), according to the manufacturer's instructions, and diluted 1:2 in nuclease-free water for qRT-PCR. All qRT-PCR was performed using the TaqMan Universal PCR Master Mix (Thermo Fisher Scientific). Each reaction consisted of 1 µl cDNA template, 5 µl master mix, 0.5 µl primers, and 3.5 µl nuclease-free water. Samples were loaded in a 384-well plate and thermocycling was performed using the Quantstudio 7 Flex system (Applied Biosystems, Warrington, UK) using the following thermal cycle: hold 2 min at 50 °C; hold 10 min at 95 °C; 40 cycles: 15 s at 95 °C, 1 min at 60 °C; hold at 4 °C. Data were captured and primary analyses performed using Expression Suite Software v 1.1 (Applied Biosystems) and the ΔΔCT method, using GAPDH as the housekeeping gene. Specifics of the TaqMan assays used, all of which were purchased from Thermo Fisher Scientific, are available on request.
2.6.3. Effluent analysis
Nitric oxide (NO) release was monitored indirectly using the Greiss assay, a widely used test which measures the levels of nitrite (NO2), a stable metabolite of NO. The nitrite content of culture media was assayed in triplicate against a sodium nitrite standard curve. Commercially available enzyme-linked immunosorbent assays (ELISAs) were used to examine the release of PGE2 (KGE004B), MMP-1 (DY901B), IL−6 (D6050) and hyaluronan (HA; DHYAL0; all R&D systems, Abingdon, UK). Optimisation was performed to determine suitable dilutions for each analyte to fit within the recommended range of the corresponding assay and samples were diluted as required in culture media and assay diluent before being analysed in duplicate. For all analytes, the detection limit (DL) was determined [21]. The DL was based on the standard deviation (SD) (σ) of the response, and the slope (S) and can be expressed as follows:
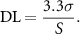
For all analytes, values below the DL were replaced with the DL value to enable statistical analysis, as is common practice.
2.7. Barrier function assessment
To assess barrier function in monoculture and co-culture chips, fluorescent tracer dye was added to the culture media in the top channel only throughout the duration of the experiment. To examine transport of differently sized molecules, cascade blue conjugated 3 kDa dextran (Thermo Fisher Scientific) and FITC-conjugated 70 kDa dextran (Sigma-Aldrich) were used at a concentration of 0.1 mg ml−1. In brief, the synovium-on-a-chip was disconnected from the automated culture module, ZÖE (Emulate), and 100 µl media removed from each reservoir. Fluorescence intensity was measured at 355 nm and 485 nm and the concentration of dye was determined for the dosing channel (top channel) and receiving channel (bottom channel) using a standard curve. This data was used to assess the apparent permeability (Papp) according to the following calculation:
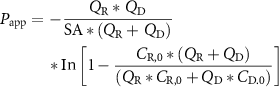
where Papp is the apparent permeability in units of cm s−1, SA is the surface area of the co-culture channel (0.17 cm2), QR and QD are the fluid flow rates in the receiving and dosing channels, respectively, in units of cm3 s−1, and CR,0 and CD,0 are the recovered concentrations in the receiving and dosing channels respectively (Emulate).
2.8. Histology, immunocytochemistry and confocal imaging
While all cells used in the present study were commercially sourced, we used primary synovial tissue collected from patients undergoing treatment for OA in the knee (collected for a separate ongoing study) to generate the representative images of healthy and inflamed synovium shown in figure 1(A) only. All patients provided written informed consent, approved by the National Research Ethics Committee in 11/NW/0875. Samples were formalin fixed and paraffin embedded before being sectioned at a thickness of 5 µm and stained with haematoxylin and eosin. Images were captured using a DS-FiL camera (Nikon Corporation, Tokyo, Japan).
Immunocytochemistry of hFLS monolayers and synovium chips was performed according to established protocols. In brief, samples were fixed with 4% PFA and then washed with PBS. This was followed by permeabilisation with 0.5% triton X-100 for 5 min, blocking with 5% donkey serum for 1 h at room temperature and the application of primary antibodies overnight at 4 °C in 0.1% bovine serum albumin (BSA; Sigma-Aldrich)/PBS. Primary antibodies used in this study are as follows: fibronectin-1 (ab2413, Abcam, Cambridge, UK), cadherin-11 (5B2H5, Invitrogen, Loughborough, UK), lubricin (ab28484, Abcam), VE-cadherin (sc-9989, Santa Cruz Biotechnology, California, USA), platelet endothelial cell adhesion molecule-1 (PECAM-1)/CD31 (ab9498, Abcam), intercellular adhesion molecule-1 (ICAM-1) (ab2213, Abcam). After 24 h samples were washed with 0.1% BSA/PBS and incubated with appropriate Alexa Fluor-conjugated secondary antibodies (Molecular Probes, Massachusetts, USA) for 1 h at a dilution of 1:500. Nuclei were counterstained with DAPI (D3571, Thermo Fisher Scientific) and actin labelled with Alexa Fluor 555-conjugated Phalloidin (Santa Cruz Biotechnology) for 1 h at room temperature. Primary antibodies were diluted at 1:200, secondary antibodies at 1:500 and DAPI was used at 1 ug ml−1. Membranes were mounted cell side up with Fluoromount G (00-4958-02, Thermo Fisher Scientific). Samples were imaged using a Nikon CSU-W1 SoRa Spinning Disk Confocal (Nikon) using a CFI Plan Fluor 10× objective (0.3 NA). Isotope controls are not included for immunocytochemistry as our group has extensive experience in the use of these optimised antibodies which have limited non-specific activity and label characteristic structures which are extremely unlikely to be shown by non-specific binding.
2.9. Statistics and data analysis
Statistical analysis was performed using GraphPad Prism version 9.5 for Windows (GraphPad Software, San Diego, California USA). Prior to statistical analysis, all data was assessed for normality (Shapiro–Wilks test). With normal distribution confirmed, statistical significance was assessed by Students t-test, one-way, two-way or three-way ANOVA followed by Sidak's multiple comparisons test, as highlighted in the individual figure legends. The threshold for statistical significance was set at 0.05. Data is represented as mean ± SD. Statistical significance is displayed as p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***, p < 0.0001 = ****. Data from multiple donors are displayed in unique colours which remain consistent throughout all figures.
3. Results
3.1. Generation of a vascular channel for synovium-on-a-chip
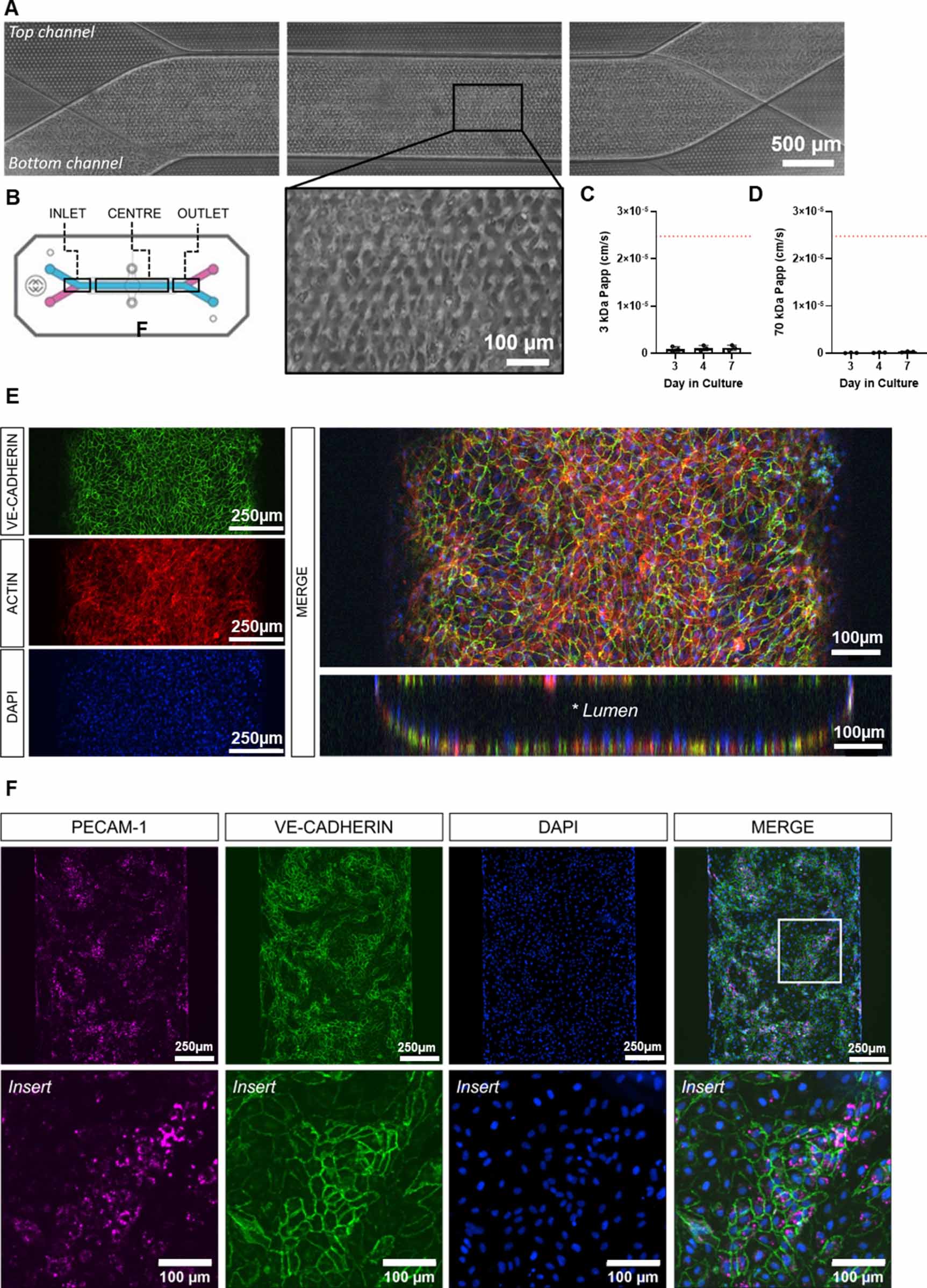
HUVECs were seeded into the bottom channel of the chip S1® and cultured for 48 h prior to the addition of hFLS. HUVECs attached well to ECM coated surfaces, forming an even monolayer through the length of the channel (figure 2(A)). An observable permeability barrier was present from day 3 in culture following connection to the automated culture module and application of fluid flow, which acted to reduce diffusion between the channels (figures 2(C) and (D)). Tight junction formation was observed with VE-Cadherin labelling (figures 2(E) and (F)) and cells labelled positive for PECAM-1; figure 2(F). HUVECs grew on all inner surfaces of the bottom channel forming a tubule-like structure with a hollow lumen (figure 2(E)).

Figure 2. Generation of a vascular channel within the organ-chip system using human umbilical vein endothelial cells (HUVECs). (A) Brightfield images of HUVECs evenly seeded within the bottom channel of the Chip S1®. (B) Schematic of the Chip S1® identifying the areas imaged in figure (A). Barrier function assessment of HUVECs monoculture chips to determine permeability to (C) 3 kDa dextran and (D) 70 kDa dextran under flow (30 µl h−1, n = 3 chips). Statistical analysis: one-way ANOVA with Sidak's multiple comparisons. The dotted red line indicates permeability in cell-free organ-chips. (E) Representative images of HUVECs monoculture chips labelled for VE-cadherin (green), actin (red) and nuclei (blue). HUVECs proliferate around the internal surface of the channel forming a tubule-like structure, highlighted in the reconstructed Z projection. (F) Representative images of HUVECs monoculture chips co-labelled for PECAM-1 (magenta) and VE-cadherin (green) demonstrating robust tight junction formation. Nuclei are counterstained with DAPI (blue).
Download figure:
Standard image High-resolution image3.2. Generation of synovial lining component of synovium-on-a-chip
Following HUVECs pre-culture, healthy hFLS were seeded into the top channel of the chip S1® (figure 3(A)). hFLS attached well along the length of the channel (figure 3(A)) and could be successfully cultured for 24 h without media refreshment with no negative effects on cell viability. Matrigel™ has commonly been used for the generation of synovial organoids [8, 22, 23]. However, within the chip environment, hFLS were found to exhibit a more invasive phenotype when cultured on Matrigel. Cells migrated through the membrane pores within 24 h of seeding and grew on the opposing side of the membrane (figures S1(B) and (C)). Therefore, initial studies explored the use of alternative ECM coatings to limit migration, with membrane coatings optimised using transwell cultures (figure S2), in which collagen type I was found to be most effective at limiting/delaying this behaviour (figures S2(B) and (C)).

Figure 3. Generation of a vascularised synovium-on-a-chip using healthy human fibroblast-like synoviocytes. (A) Brightfield images of co-culture chips with HUVECs seeded within the bottom channel of the Chip S1® and hFLS in the top channel (day 7). (B) Schematic of the Chip S1® identifying the areas imaged in figure 2(A). Barrier function assessment of co-culture chips to determine permeability to (C) 3 kDa dextran and (D) 70 kDa dextran under flow (30 µl h−1, n = 6 chips, N = 2 donors). Statistical analysis: one-way ANOVA with Sidak's multiple comparisons. The dotted red line indicates permeability in cell-free organ-chips. Representative images of hFLS in co-culture chips labelled for synoviocyte markers (E) fibronectin-1, (F) cadherin-11 or (G) lubricin (green). In all images, actin and nuclei are counterstained with phalloidin (red) and DAPI (blue) respectively.
Download figure:
Standard image High-resolution imageOn collagen type I coated surfaces, hFLS formed a loose network at the surface of the membrane (figures 3(E) and (F)). Co-culture chips formed a robust permeability barrier to 70 kDa dextran but were found to be more permeable to 3 kDa dextran than monoculture chips (figures 3(C) and (D)). Deposition of fibronectin-1 (FN-1) was observed in the synovial compartment (figure 3(E)) and hFLS exhibited positive labelling for PRG4 (lubricin), which was found distributed throughout the cell cytoplasm (figure 3(G)). Furthermore, expression of the synovial marker cadherin-11 could be observed at points of cell–cell contact, overlapping with actin labelling (figure 3(F)). In addition, hFLS-secreted HA was detected in the culture media (figure 6(F)).
3.3. Optimisation of mechanical loading regimes using 2D cyclic stretch
FLS exhibit mechanosensitive gene expression of HA synthase enzymes (HAS-1, 2 and 3) and production of HA both in vivo [24, 25] and in vitro [26]. However, many of these studies use cells from animal models, cell lines or cells isolated from diseased tissue. We therefore examined this response in a 2D stretch system on healthy human FLS, prior to testing in the chip environment. Isolated hFLS were cultured on collagen type I coated elastomeric membranes and subjected to cyclic tensile strain at 2% or 12% strain for 2–24 h. We found that after 24 h synoviocytes with strain began to detach from the membrane even at the lowest strain levels (data not shown), therefore a shorter loading regime of 2 h was used. Actin stress fibre formation was observed in hFLS subjected to 12% strain while only faint actin labelling was observed under both control (0% strain) and 2% strain conditions (figure 4(A)). Significant upregulation of HAS-1 (p = 0.0087) and HAS-3 (p = 0.0112) were observed in response to 12% strain only (figures 4(B) and (D)). HAS-2 gene expression was found to be significantly downregulated by 2% strain (p = 0.0013), but not 12% strain (figure 4(C)). Gene expression of HAS-2 and PRG-4 (encoding lubricin) were not significantly altered by the application of strain (figures 4(C) and (E)).
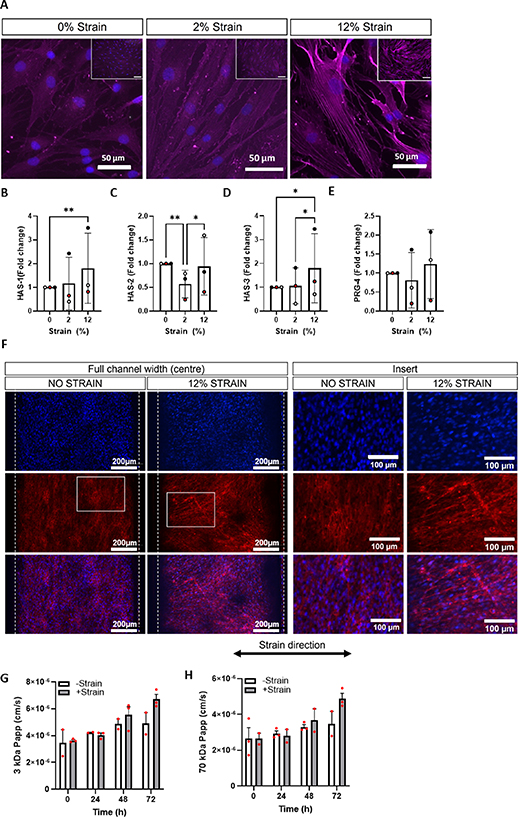
Figure 4. Fibroblast-like synoviocytes sense mechanical stimulation and exhibit mechanosensitive changes in gene expression. Note: figures (A)–(E) were generated using isolated hFLS in 2D monocultures in the Flexcell® system, subjected to 0%, 2% or 12% uniaxial cyclic tensile strain for 2 h at 0.2 Hz. Figures (F)–(H) were generated using the organ-chip system, in which cells were subjected to 12% uniaxial cyclic tensile strain for 2 h at 0.1 Hz. (A) Representative images of hFLS labelled for actin (magenta) to highlight stress fibre formation, nuclei are counterstained with DAPI (blue). Gene expression levels for (B) HAS-1, (C) HAS-2, (D) HAS-3 and (E) PRG-4 were examined immediately after mechanical stimulation. Gene expression levels were normalised to GAPDH and expressed as a fold change relative to the 0% strain control using the ΔΔCt method (n = 3 samples, N = 3 donors). Statistical analysis: two way ANOVA with Sidak's multiple comparisons, * = p < 0.05 and **p = <0.01. Synovium-on-a-chip was cultured for 7 d then subjected to mechanical stimulation (2 h, 12% strain at 0.2 Hz) for 72 h. (F) Representative images of hFLS labelled for actin (red) in the central region of the channel (peak stress), nuclei are counterstained with DAPI (blue). Barrier function assessment of permeability to (G) 3 kDa and (H) 70 kDa fluorescent dextran in co-culture model (n = 3 chips, N = 1 donor). Statistical analysis: two way ANOVA with Sidak's multiple comparisons.
Download figure:
Standard image High-resolution imageWe then applied these findings to co-culture organ-chips for further optimisation. In unstrained chips, actin labelling of hFLS was weak, with little definition of stress fibre formation, and cells had a strong tendency to align along the length of the channel within a loose network (figure 4(F)). Upon the addition of 12% strain, this cell alignment was disrupted, and cells stained more intensely for actin, although stress fibre formation was not easily observable under the culture conditions (figure 4(F)). There was no significant difference in barrier permeability to 3 kDa or 70 kDa dextran in response to strain (figures 4(G) and (H)). Consistent with our observations of increased HAS gene expression in response to stretch, we observed a trend towards an increase in HA concentration in the conditioned media from the synovium channel in response to strain, but this was not statistically significant (data not shown).
3.4. Interleukin−1β (IL−1β) promotes proinflammatory signalling and release of catabolic factors in 2D cultures in the presence of strain
The application of IL−1β to hFLS was used to simulate synovial inflammation. The response to IL−1β was initially examined in 2D on tissue culture plastic and, subsequently, in the presence of strain. Isolated hFLS were subjected to IL−1β treatment 0–10 ng ml−1 for 24 h to identify an optimal dose for use in subsequent studies, and culture supernatant examined for the presence of inflammatory signalling molecules. No significant alterations in the release of nitric oxide (NO) were observed in response to IL−1 (figure S3(A)). However, significant release of PGE2 was observed at all concentrations of IL−1β compared to the control condition, and release of IL−6 observed from 1 ng ml−1 (figures S3(B) and (C)). Significant release of the catabolic enzyme MMP-1 was also observed from 1 ng ml−1 IL−1β (figure S3(D)). No significant effects on cell viability were observed but the FLS began to look stressed morphologically upon the addition of 10 ng ml−1 (data not shown). For this reason, an IL−1β dose of 1 ng ml−1 was used in subsequent experiments. Gene expression was also analysed following the addition 1 ng ml−1 IL−1β to 2D hFLS cultures. There was a trend towards increased expression of catabolic markers ADAMTS-4, ADAMTS-5, and MMP-3 while MMP-1 expression was significantly increased (p = 0.0006, figure 5(B)). Moreover, expression of MMP-13 was observed in cells treated with IL−1β but not untreated cultures (not shown).
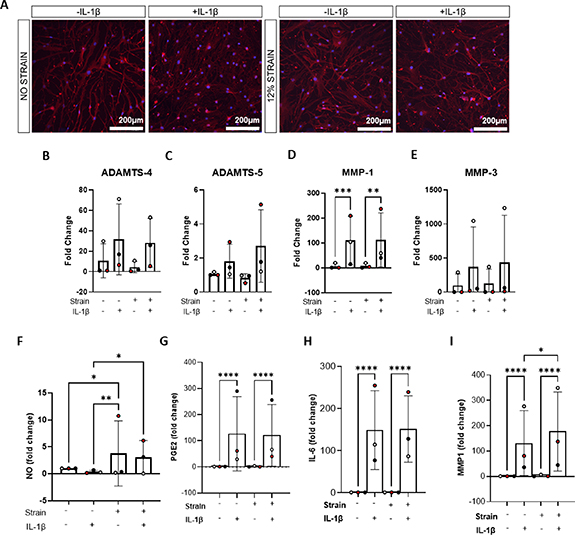
Figure 5. IL−1β promotes pro-inflammatory signalling and the release of catabolic factors in 2D culture. Isolated hFLS were cultured in 2D monocultures for 22 h then subjected to 0% or 12% cyclic tensile strain using the Flexcell ® for 2 h at 0.2 Hz in the presence or absence of 1 ng ml−1 IL−1β (24 h treatment total). (A) Representative images of hFLS labelled for actin (red) and nuclei counterstained with DAPI (blue). Note that figures (B)–(E) show data generated by qRT-PCR and figures (F)–(I) data generated by ELISA. Gene expression levels for (B) ADAMTS-4, (C) ADAMTS-5, (D) MMP-1 and (E) MMP-3 were examined immediately after mechanical stimulation. Gene expression levels were normalised to GAPDH and expressed as a fold change relative to the 0% strain control using the ΔΔCt method (n = 3 samples, N = 3 donors). Culture supernatants were collected at the end of mechanical stimulation and examined for (F) nitric oxide (NO) (G) prostaglandin E2 (PGE2) (H) interleukin-6 (IL−6) and matrix metalloproteinase-1 (MMP-1) content (n = 3 samples, N = 3 donors). Statistical analysis: two way ANOVA with Sidak's multiple comparisons, * = p < 0.05, **p = <0.01, *** = p < 0.001 and **** = p < 0.0001.
Download figure:
Standard image High-resolution imageThe hFLS response to 1 ng ml−1 IL−1β was then explored in the presence of 12% cyclic tensile strain, using the optimised regime based on the previously mentioned experiment. There was a significant upregulation of PGE2 in the presence of IL−1β in both the unstrained (+IL−1β: 2834.59 pg ml−1 ± 3272.13SD vs −IL−1β: 22.83 pg ml−1 ± 19.72SD; p < 0.0001) and strained (+IL−1β: 2871.57 pg ml−1 ± 2706.58SD vs −IL−1β: 42.96 pg ml−1 ± 56.81SD; p < 0.0001) conditions (figure 5(G)). Similar trends were observed in the secretion of IL−6, in response to IL−1β addition, in the strained (+IL−1β: 17 774.44 pg ml−1 ± 9469.82SD vs −IL−1β: 118.48 pg ml−1 ± 3.14SD; p < 0.0001) and unstrained conditions (+IL−1β: 17 472.40 pg ml−1 ± 9603.14SD vs −IL−1β: 114.43 pg ml−1 ± 0SD; p < 0.0001; figure 5(H)). MMP-1 was similarly upregulated in the presence of IL−1β in the strained (+IL−1β: 3914.25 pg ml−1 ± 4007.83SD vs −IL−1β: 31.15 pg ml−1 ± 32.12SD; p < 0.0001) and unstrained conditions (+IL−1β: 3079.46 pg ml −1 ± 3270.75SD vs −IL−1β: 71.55 pg ml−1 ± 107.40SD; p < 0.0001; figure 5(I)). Interestingly, MMP-1 secretion was further upregulated in the presence of strain and IL−1β, compared to IL−1β alone (+IL−1β, +strain: 3914.25 pg ml−1 ± 4007.83SD vs +IL−1β, −strain: 3079.46 pg ml−1 ± 3270.75SD; p = 0.0465; figure 5(I)). NO secretion was significantly greater with the application of strain, both in the presence of IL−1β (+IL−1β, +strain: 9.00 µM ± 12.83SD vs +IL−1β, −strain: 2.40 µM ± 5.45SD; p = 0.0207) and in the absence of IL−1β (−IL−1β, +strain: 17.89 µM ± 24.78SD vs −IL−1β, −strain: 9.28 µM ± 11.46SD; p = 0.0256),although this was largely due to the increased secretion detected in the hFLS of one of the three donors (red; figure 5(F)). Gene expression analysis of hFLS in the presence of CTS also revealed similar trends following the addition of IL−1β, with upregulation ADAMTS-4 and -5, MMP-3 and MMP-1, in which the difference was statistically significant (p = 0.0023; figure 5(B)).
3.5. Interleukin-1β triggers robust release of pro inflammatory mediators and catabolic enzymes in synovium-on-a-chip
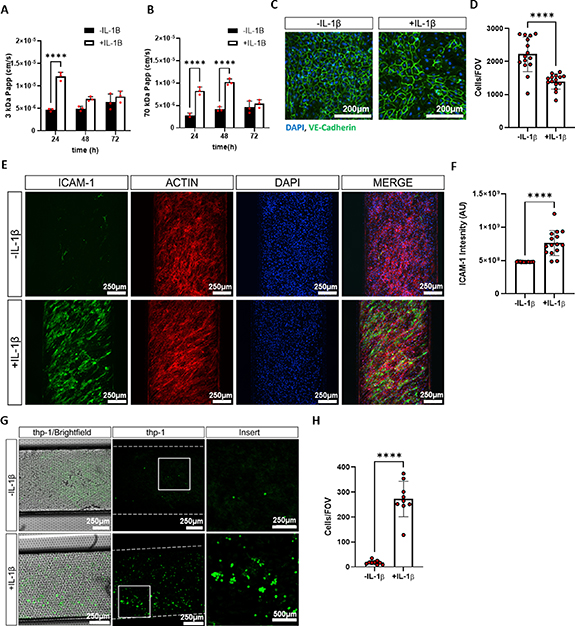
The response of hFLS to inflammatory stimuli in the vascularised synovium-on-a-chip was then examined by the application of 1 ng ml−1 IL−1β to the top channel for three days from day 7. There was no observable difference in cell distribution or actin structure after 72 h of IL−1β treatment (figure 6(A)). After 24 h, there was a significant increase in barrier permeability to 3 kDa dextran. Studies in monoculture chips (hFLS only or HUVECs only) suggested that this response was due largely to alterations in the vascular channel, with a similar reduction in barrier function observed in HUVECs monoculture chips at 24 h, which then recovered (figure S4). Consistent with this finding, VE cadherin labelling was found to be more variable in some regions after 72 h of IL−1β treatment, and cell number was reduced. Interestingly, permeability to 70 kDa dextran was also increased and remained high at 48 h post stimulation with IL−1β. However, this response was not seen in HUVECs monoculture chips, suggesting that the effect was mediated, at least in part, by the hFLS. A functional barrier was restored by 48 h with respect to 3 kDa dextran, however chips remained permeable to 70 kDa dextran until 72 h post application of IL−1β.

Figure 6. Inflammatory responses could be recapitulated in the vascularised synovium-on-a-chip. Synovium-on-a-chip subjected to IL−1β stimulation (1 ng ml−1) for a total of 72 h in the presence of mechanical stimulation (2 h, 12% strain at 0.2 Hz every 22 h). (A) Representative images of hFLS labelled for actin (red) in the central region of the channel (peak stress), nuclei are counterstained with DAPI (blue). Chip effluent was collected at the end of the mechanical stimulation period (24, 48 and 72 h) and examined for the release of (B) NO (n = 3 chips, N = 3 donors) (C) PGE2 (n = 3 chips, N = 3 donors) (D) IL−6 (n = 3 chips, N = 3 donors) (E) MMP-1 (n = 3 chips, N = 3 donors) (F) HA (n = 1 chip, N = 3 donors). Statistical analysis: three way ANOVA with Sidak's multiple comparisons, * = p < 0.05, **p = <0.01, *** = p < 0.001 and **** = p < 0.0001.
Download figure:
Standard image High-resolution imageThe secretion of the aforementioned pro-inflammatory and catabolic mediators, as well as the SF constituent, HA, were assessed in the conditioned media collected from the hFLS channel at 24 h, 48 h and 72 h after the addition of IL−1β. There were no significant differences observed in terms of NO release in the presence or absence of IL−1β (figure 6(B)). At all three timepoints, increased secretion of PGE2 was observed in the presence of inflammatory stimuli, which was statistically significant at 72 h (+IL−1β: 11 207.86 pg ml−1 ± 4427.86SD vs −IL−1β: 22.52 pg ml−1 ± 26.05SD; p = 0.0039; figure 6(C)). The secretion of IL−6 was significantly higher in the IL−1β treated chips compared to the control chips at 24 h (+IL−1β: 13 735.70 pg ml−1 ± 7205SD vs −IL−1β: 177.34 pg ml−1 ± 136.92SD; p = 0.0004), 48 h (+IL−1β: 12 677.76 pg ml−1 ± 7555.83SD vs −IL−1β: 160.3 pg ml−1 ± 44.05SD; p = 0.0009) and 72 h (+IL−1β: 9156.36 pg ml−1 ± 4626.15SD vs −IL−1β: 153.49 pg ml−1 ± 0SD; p = 0.0318). At 24 h, the secretion of MMP-1 was significantly lower in the control chips (2347.33 pg ml−1 ± 1648.66SD) compared to in those to which IL−1β was added (3792.27 pg ml−1 ± 1551.08SD; p = 0.0176), but this difference was lost at the later timepoints (figure 6(E)). The secretion of HA was also stimulated in the synovium chips by the addition of IL−1β and was found to be higher in the IL−1β stimulated chips compared to the control chips at all three timepoints, with statistical significance at 48 h (+IL−1β: 1820.36 ng ml−1 ± 936.06SD vs −IL−1β: 377.86 ng ml−1 ± 0SD; p= 0.0249) and 72 h (+IL−1β: 2019.56 ng ml−1 ± 831.01SD vs −IL−1β: 377.86 ng ml−1 ± 0SD; p = 0.01; figure 6(F)).
3.6. Endothelial activation is observed in synovium-on-a-chip in response to IL−1β
The expression of ICAM-1 was increased within the endothelial channel of synovium-on-a-chip, and labelling intensity was significantly higher, in IL−1β treated chips versus untreated control chips (figures 7(E) and (F)). No positive labelling of ICAM-1 was observed in the monoculture inlet region of the chip where there was no mixing between synovium and vascular channels (figure S5), suggesting that IL−1β, or one of its downstream mediators is likely responsible for this effect.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Synovial inflammation promotes endothelial cell activation and attachment of THP-1 monocytes in the vascularised synovium-on-a-chip. Synovium-on-a-chip subjected to IL-1β stimulation (1 ng ml−1) for a total of 72 h in the presence of mechanical stimulation (2 h, 12% strain at 0.2 Hz every 22 h). Chip effluent was collected at the end of the mechanical stimulation period (24 h) and barrier function assessment performed to determine permeability to (A) 3 kDa and (B) 70 kDa fluorescent dextran in the co-culture model (n = 3 chips, N = 1 donor). Statistical analysis: two way ANOVA with Sidak's multiple comparisons, **** = p < 0.0001. (C) Representative images of HUVECs labelled for VE-cadherin (green) and nuclei counterstained with DAPI (blue) to observe tight junction formation in the endothelium. (D) Quantification of cell number in the endothelium (n = 15 fields of view (FOV), N = 1 donor). Statistical analysis: students t-test, **** = p < 0.0001. (E) Representative images of endothelial cells in synovium-on-a-chip labelled for ICAM-1 (green), actin (red) and nuclei counterstained with DAPI (blue). (F) Quantification of ICAM-1 expression (n = 15 fields of view (FOV), N = 1 donor). Statistical analysis: students t-test, **** = p < 0.0001. THP-1 monocytes were labelled with a fluorescent tracker then added to the bottom channel of synovium-on-a-chip at the end of mechanical stimulation period and cell adhesion assessed by confocal microscopy. (G) Representative images of THP-1 cell adhesion (green) to the underside of the membrane (Brightfield) (H) Quantification of cell adhesion (n = 15 fields of view (FOV), N = 1 donor). Statistical analysis: students t-test, **** = p < 0.0001.
Download figure:
Standard image High-resolution image{kind=link}
3.7. Monocyte recruitment is increased in the inflammatory model of synovium-on-a-chip
To determine if the activated endothelium (figures 7(E) and S5) is functionally relevant in synovium-on-a-chip, THP-1 monocytes were introduced to the bottom channel of the chip. Monocytes were labelled with cell tracker and incubated in the bottom channel under static conditions for 3 h before the channel was flushed with fresh medium at 1000 µl h−1. Few cells attached to the surface of the endothelial channel in untreated chips, however in IL−1β treated chips, attachment was significantly increased by 16-fold (p < 0.0001; figures 7(G) and (H)).
4. Discussion
The critical role of the human synovium in maintaining joint homeostasis, and the corresponding contribution of synovitis to the onset and progression of arthritic joint disease, makes this tissue a promising target for the development of novel therapeutics. However, there is an unmet need for widely available, physiologically relevant in vitro models of synovium that accurately predict in vivo responses. The present study sought to address this deficiency by generating a microfluidic-based, human synovium-on-a-chip that emulated several features of the healthy, and inflamed synovium, on a background of biomechanical stimulation. The human vascularised synovium-on-a-chip was developed using a commercially available chip (Chip-S1®) and accompanying hardware (Emulate Inc.). There are benefits and drawbacks of both in-house and commercial OOAC systems, depending on factors such as the application, user need, reproducibility, number of replicates, cost and availability. A commercially available organ-chip system was selected in this case to allow for the wider research community to adopt the optimised model without the need for specialist knowledge on chip fabrication techniques.
In vivo, the synovium can be divided into two regions: the intima, or synovial lining, which interfaces with the SF-filled joint cavity, and the more fibrous subintima beneath, which merges with the joint capsule [27]. We demonstrated that hFLS, cultured in synovium-chip, exhibited behaviour that was characteristic of the intima, or lining layer, of native human synovium. In this way, hFLS formed a monolayer within the top channel of the synovium-chip, establishing a loose network and depositing fibronectin, a key component of the synovial ECM, the maintenance of which FLS are responsible for in vivo. Additionally, hFLS demonstrated positive immunolabelling of cadherin-11, a cell adhesion molecule essential for maintaining the normal synovial architecture, by influencing cellular organisation and ECM development. The importance of this protein in the development and organisation of the synovium has been previously demonstrated using a cadherin-11 deficient mouse model in which the synovia exhibited marked hyperplasia and a lack of a compact lining layer of cells [28]. Further, FLS isolated from this mouse model fail to replicate the synovial lining layer arrangement, compared to FLS isolated from wild-type mice.
In addition to the maintenance of the synovial ECM, a primary function of the synovium is regulation of the volume and composition of the SF. This role is partly performed through the biosynthesis of constituent lubricant proteins by the resident FLS. In the synovium-on-a-chip, hFLS emulated this behaviour, staining positively for lubricin and demonstrating the secretion of HA, two of the major SF constituents. Additionally, in vivo, the combined barrier function of the semi-permeable synovial and endothelial membranes acts to simultaneously concentrate blood plasma proteins by filtration into the SF and prevent the loss of FLS-secreted lubricants from the joint space [29]. In the organ-chip system, the barrier function of the individual endothelial and synovial components, as well as the co-culture, were shown to reduce the diffusion of labelled dextran from the top to the bottom channel, compared to a cell-free chip S1®. The molecular weight of HA in normal SF ranges from 6000–10 000 kDa [30], and lubricin from 200–400 kDa [29]. Therefore, the dextran used for barrier function analyses in the present study was considerably smaller than synovium-secreted SF constituents. However, the synovium is a size-selective barrier, and its permeability has previously been reported to be inversely proportional to molecular size [31, 32]. Therefore, larger proteins, such as SF constituents secreted by the hFLS, should theoretically be conserved in the synovium channel. Critically, while the synovial and endothelial barriers reduced the diffusion of both 3 kDa and 70 kDa dextran, their diffusion was not completely prevented suggesting that smaller proteins such as albumin (∼67 kDa), which are filtered from the blood plasma [33] would be able to pass across the barrier in some capacity. The lack of testing of larger proteins represents a limitation of the present study and future research will include the exogenous addition of such plasma proteins to investigate their filtration within the model and more robustly characterise the barrier and filtration properties of the model system.
Both the healthy and inflamed vascularised synovium models were developed on a background of mechanical stimulation (fluid shear and cyclic tensile strain). There is widespread evidence that mechanical stimulation regulates cell behaviour including the response to anti-inflammatories and other potential therapeutics. It is therefore appropriate to include this aspect of the cellular environment within organ-chip models such as this (For review see Thompson et al, 2020 [34]). In the present study, we demonstrated that 0%–12% cyclic tensile strain applied to hFLS for 2 h at 0.2 Hz, is sufficient to upregulate the expression of genes, HAS-1 and HAS-3, which are associated with synthesis of the SF component, HA (figure 4). This physiological loading regime was therefore applied within the organ-chip although it was not the aim of the present study to examine the mechanosensitive behaviour of this model. The native human synovium is highly vascularised, with capillaries commonly located just below the synovial intima [35], a feature which was replicated in the synovium-chip by the positioning of the top synovium channel and bottom vascular channel. We were able to generate a functional blood vessel element, with tuneable fluid shear and tensile strain, demonstrating characteristic matrix protein deposition and barrier function. It is well-established that the vascular system contributes significantly to pharmacokinetics and therefore the system could be utilised for drug delivery studies [36]. Additionally, increased angiogenesis is a common feature of RA and OA, and it has been established that angiogenic mediators, produced by various cell types, activate endothelial cells, which in turn release proteolytic enzymes [37]. Further, vasculitis is prevalent in RA, and has been implicated in the development of the systemic features of the disease [38, 39]. There is further potential to model these pathogenic changes in synovium-on-a-chip through the addition of angiogenic (e.g. vascular endothelial growth factor) and inflammatory mediators (e.g. IL−1β) to the bottom channel. Examining the behaviour of the endothelial channel and its interaction with the synovium channel under these conditions may improve understanding of disease onset and progression and highlight further drug targets. Both intraarticular (e.g. corticosteroids) and intravenous (e.g. tumour necrosis factor-inhibitors, B-cell inhibitors) therapies are used in the treatment of RA, and the inclusion of both a synovial and vascular element in our model offers the possible opportunity to test therapeutics designed for both treatment routes.
Dysregulation of synovial function is implicated in the onset and progression of arthritic joint diseases, in which synovial inflammation (synovitis) is common. In the current study, IL−1β was used to mimic synovial inflammation, resulting in the increased secretion of inflammatory (PGE2, IL−6) and catabolic (MMP-1) mediators by hFLS within synovium-on-a-chip. It is widely reported that, in vivo, these mediators contribute to the degeneration of articular cartilage, the hallmark feature of arthritic diseases [40]. Recapitulating this response to inflammatory insult is crucial for physiologically relevant models of disease. The authors are keen to explore the mechanisms by which these mediators affect articular cartilage and will advance the model further to include a cartilage element. Upon the application of an inflammatory stimulus, an increase in HA secretion was observed, corroborating in vitro findings by Stefani and colleagues who showed that HA secretion from explant and tissue engineered bovine synovium was increased upon the application of IL−1α [8]. Interestingly, these authors also reported that the average molecular weight of secreted HA was decreased following the application of inflammatory stimuli, thought to be due to the increased breakdown of larger HA molecules, but this was not investigated in the present study.
The application of an inflammatory stimulus to the synovial channel also led to an increase in the permeability of the barrier between the two channels of the chip, to both 3 kDa and 70 kDa dextran. Our results indicated that the change in barrier function in the co-culture chips was largely due to alterations in the vascular channel, with changes in permeability of the endothelial channel-only monoculture chip mirroring that of the co-culture chip. It is well-established that, in vivo, vascular permeability is increased at sites of inflammation and that this phenomenon is mediated by the activation of the endothelium by inflammatory cytokines, such as IL−1β [41, 42] thus the synovium-on-a-chip is able to mimic this physiological process.
Local activation of the endothelium is also implicated in the recruitment of circulating inflammatory cells to sites of tissue inflammation [43]. Endothelial cell activation leads to the increased expression of cell adhesion molecules and subsequently to increased endothelial–leukocyte interactions [14, 44]. A hallmark feature of synovial inflammation is an increase in macrophage numbers, which arise from circulating monocytes recruited to areas of synovial inflammation, and which subsequently differentiate into macrophages in situ [45, 46]. Monocyte extravasation from the circulation into the synovial tissue is a chemokine-driven, multi-step process which begins by weak adhesion to endothelial cell selectins within 1–2 h, followed by firm adhesion to V-CAM1 and ICAM-1 [47, 48]. In the synovium-chip, we were able to demonstrate that after 3 h there was increased attachment of monocytes to the surface of the endothelial channel in the inflamed condition, thus mimicking the early stages of inflammatory cell recruitment seen in vivo. However, this experiment was carried out using only one chip per condition and cells from only one donor, as a very early proof-of-concept. Additionally, we only examined monocyte behaviour over a period of 3 h, under static conditions, representing a further limitation of the present study. Future work will seek to more robustly characterise monocyte behaviour in the system, including migration between channels.
Of note, in the present study, a monocytic cell line (THP-1), rather than primary monocytes, was used to investigate monocyte attachment to the endothelium in the synovium-on-a-chip model. The THP-1 cell line is widely used in the development of in vitro models and reliably replicates primary monocyte function (for review, see Chanput et al 2014 [49]). Future patient stratification studies are developing a personalised version of the organ-chip to examine donor variability using primary cells from synovial biopsies (Versus Arthritis Fellowship #22876). However, donor matched endothelial cells and monocytes are difficult to routinely obtain. Therefore, to facilitate these and other future studies and ensure that donor variability could be attributed to the behaviour of the hFLS, the source of endothelial cells and monocytes was standardised.
Throughout the analyses performed in the present study, there was clear variation between donors, indicating that while the synovium-chip is reproducible (within-donor, between-chip variability was relatively low), we were able to maintain donor variability in the system. This feature is currently being exploited for the development of disease-stratified and even personalised synovium-chips, using cells isolated from characterised, primary human tissues.
5. Conclusions
This study presents the first, human, vascularised synovium-on-a-chip model, built upon a commercially-available platform, which can be used to understand normal synovial function and pathogenic dysfunction. The model incorporates physiological mechanical loading and successfully recapitulates key features of native synovium biology. This new model offers a versatile, translatable platform which can easily be adopted to examine human synovial biology and inflammation, identify novel druggable targets and potentially test new therapeutics.
Acknowledgments
We would like to express our gratitude to our funders (Versus Arthritis, Orthopaedic Institute Ltd., MRC & EPSRC) and our colloborators who have made this work possible. Timothy Hopkins is funded by a Versus Arthritis Foundation Fellowship (22876). This work forms part of the research portfolio of the National Institute for Health Research Barts Biomedical Research Centre (NIHR203330).
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Conflict of interest
Competing Interests and Funding: M M K is Director, and C L T was formerly the Centre Scientist, of the Queen Mary + Emulate Organs-on-Chips Centre, which is part funded by Emulate Inc. Emulate Inc. were not involved in the preparation of this manuscript. The remaining authors declare that they have no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Ethical Statement
The study was conducted according to the guidelines of the Declaration of Helsinki. Favourable ethical approval for the generation of tissue histological figures was given by the National Research Ethics Service (11/NW/0875, 2012).
Funding
UK Research and Innovation: Engineering and Physical Sciences Research Council. EP/NO27264/1.
UK Research and Innovation: Medical Research Council. MR/R02569X/1 & MR/T015462/1.
Orthopaedic Institute Limited. RPG190.
Versus Arthritis. 22876
Supplementary data (0.5 MB PDF)